TÍTULO: IMPLEMENTAÇÃO DE UM MODELO COMPUTACIONAL DE GÁS IDEAL MONOATÔMICO UTILIZANDO DINÂMICA MOLECULAR
AUTORES: Silva, J.R. (LCCV/CTEC/UFAL) ; Lira, W.W.M. (LCCV/CTEC/UFAL) ; Cintra, D.T. (LCCV/CTEC/UFAL)
RESUMO:Neste trabalho desenvolve-se um simulador numérico simplificado que utiliza o
método de Dinâmica Molecular (DM) para estudar o comportamento termomecânico de
um
sistema químico de um gás ideal em um espaço bidimensional unitário. A simulação
é
feita considerando átomos como partículas, o método de integração de Velocity-
Verlet e a função potencial de Lennard-Jones para simular os campos de energia e
calcular as forças entre os átomos considerando um raio de interação definido.
Com
o simulador desenvolvido é possível observar características peculiares de um
sistema monoatômico, como a influência de temperatura e concentração no
movimento
dos átomos. Observa-se, ainda, que a função de Lennard-Jones é uma boa escolha
para
modelagem de sistemas químicos por sua simplicidade.
PALAVRAS CHAVES: Dinâmica Molecular; Potencial deLennard-Jones; Elementos Discretos
INTRODUÇÃO:Em muitas aplicações da engenharia é interessante utilizar meios particulados
para resolver problemas complexos de interesse, podendo envolver, por exemplo, o
fenômeno de fragmentação. A abordagem pelo Método dos Elementos Discretos
(CUNDALL & STRACK, 1979) para esta categoria de problemas é apropriada em
virtude de sua natureza inerentemente descontínua. Na engenharia química, o
estudo de Dinâmica Molecular para modelagem de sistemas químicos pode ser
interessante para interpretar e até prever o comportamento de algum mecanismo
interação entre átomos, como o princípio de exclusão de Pauli e as forças de
dispersão de van de Waals.
O método de Dinâmica Molecular foi proposto inicialmente por (ALDER &
WAINWRIGHT, 1959) e é usado para modelar o comportamento de sistemas químicos
usando equações de movimento e funções que simulem campos de energia envolvidos
na interação entre espécies químicas. A dinâmica do sistema é resolvida de forma
discreta ao longo do tempo e a cada passo de integração são atualizadas
posições, velocidades e calculadas variáveis de interesse como temperatura e
energia.
A interação ocorre entre pares de átomos e possui duas características
importantes para a simulação: a) resistência à compressão; b) capacidade de
manter os átomos unidos nos estados sólido e líquido. Assim, átomos muito
próximos se repelem e, a partir de certa distância, se atraem mutuamente. A
função de Lennard-Jones é escolhida para a simulação, pois representa bem essas
características (RAPAPORT, 2004).
Dentro deste contexto, o objetivo deste trabalho é o desenvolvimento de um
simulador de Dinâmica Molecular, através do Método dos Elementos Discretos, para
observar o comportamento de um gás ideal submetido a diferentes temperaturas e
concentrações.
MATERIAL E MÉTODOS:São fornecidas as informações iniciais para a simulação (número de átomos,
tempo,
distância de interação, passo de integração e temperatura), as posições iniciais
são
estabelecidas por um parâmetro de rede e as velocidades iniciais são sorteadas
por
distribuição gaussiana.
A função de Lennard-Jones usada para átomos que estão separados por uma
distância
r,
menor que a distância de interação, é dada pela Equação 1.
V=q[(s/r)12-(s/r)6] (1)
Nessa equação, q regula a força de interação, s define uma escala de comprimento
e
r
é a distância entre os átomos. Para simplificar equações e evitar
problemas do tipo underflow e overflow, adota-se unidades adimensionais, usando
m=1, q=1 e r=rs (SABINO et al., 2009). Assim, a função é reescrita:
V=q[(1/r)12-(1/r)6] (2)
O cálculo das forças sobre cada átomo é feito com a Equação 3:
F=-∇V=12q[(1/r)14-(1/r)8] (3)
Para atualizar posições e velocidades usa-se o método de integração de Velocity-
Verlet (SCHERER, 2005), o qual utiliza as seguintes equações:
s(t+Δt)=s(t)+v(t)Δt+½f(t)Δt2 (4)
v(t+Δt)=v(t)+½{f(t+Δt)+f(t)}Δt (5)
O método é incrementado com um fator de normalização para que o sistema se
mantenha
a uma temperatura constante Tr. O fator é dado pela Equação 6.
vr=√(Tr/((T(t)+T(t+Δt))/2)) (6)
E implementado conforme a Equação 7.
v=(v-v')vr (7)
Onde, v é a componente da velocidade e v' é a média entre as velocidades dos
átomos
(TRAVIESSO, 1989).
Para minimizar a limitação do número de átomos usam-se condições de contorno
periódicas, repetindo o espaço de simulação em todas as direções.
Energia cinética e temperatura são simuladas usando, respectivamente:
E=1/(2N)∑v2 (8)
t=1/(dn)∑v2 (9)
sendo, d é a dimensão do modelo (d=2).
RESULTADOS E DISCUSSÃO:O modelo proposto é desenvolvido computacional no ambiente de programação
MATLAB, seguindo o esquema da figura 1. É estabelecido um modelo de dimensões
unitárias, onde as posições iniciais são definidas por um parâmetro de rede e as
velocidades iniciais são escolhidas por sorteio. A cada passo de tempo o
algoritmo atualiza posições e velocidades. Em seguida calcula-se a temperatura
do sistema e exibe-se a movimentação dos átomos ao lado do gráfico que ilustra a
temperatura em função do tempo.
É possível visualizar no modelo a interferência da concentração de partículas na
simulação. Observa-se na Figura 2 que no modelo (b) existem uma probabilidade
maior de encontrar partículas no raio de interação que no modelo (a) e,
portanto, a quantidade de forças de interação é alta também.
A influência da temperatura no sistema é clara: temperaturas altas ocasionam
altas velocidades e baixas temperaturas são responsáveis por velocidades mais
baixas. Essa característica da simulação é decorrente da relação entre
temperatura e movimento. Em temperaturas elevadas o grau de agitação das
partículas é alto. Isto significa que a velocidade das partículas é maior, logo
em um sistema de maior temperatura o grau de desordem das partículas é maior,
conforme pode ser visto na Figura 2 os modelos (c) e (d).
Figura 1
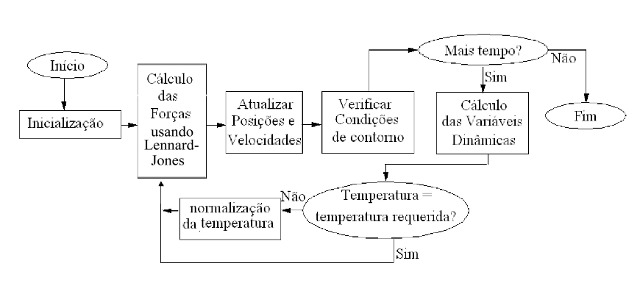
Fluxograma dos passos da simulação de Dinâmica
Molecular.
Figura 2
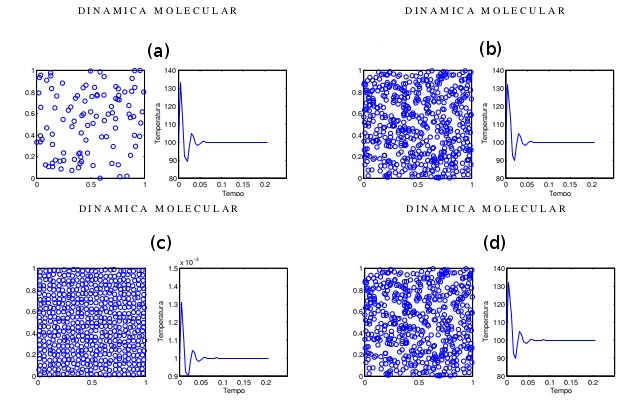
Influência da concentração e a da temperatura no
sistema. Em (a) 100 átomos e em (b) 529 átomos. Em
(c) o sistema está a 0,001K e em (d) 100K.
CONCLUSÕES:Com o trabalho foi possível concluir que o modelo bidimensional desenvolvido,
simulou muito bem um gás ideal, os diferentes parâmetros simulados forneceram
resultados satisfatórios, aproximando-se do que seria obtido em um modelo real,
onde, de fato, quanto maior a temperatura, maior o movimento entre os átomos e,
assim, maior o grau de desordem entre as partículas. Os resultados para o
parâmetro que trata da concentração (número de partículas) também se comportaram
como esperado, mostrando maior interação quando havia mais partículas no espaço
de simulação.
AGRADECIMENTOS:
REFERÊNCIAS BIBLIOGRÁFICA:ALDER, B. J.; WAINWRIGTH, T. E.. Studies in molecular dynamics. i. General method. The Journal of Chemical Physics, vol. 31, n. 2, 1959.
CUNDALL, P. A.; STRACK, O. D. L.. A discrete numerical model for granular assemblies. Geotechnique, 1979.
RAPAPORT, D. C.. The Art of molecular Dynamics Simulation. Cambridge University Press, Second Edition, 565 p., 2004.
SCHERER, C.. Métodos Computacionais da Física. Livraria da Física, 1ª Edição, 284p., 2005.
SABINO, T. L. R.; OLIVEIRA, J. P. S.; PEÇANHA, J. P.; BARROSO, J. S.; ALMEIDA, R. B.; VIEIRA, M. B.; LOBOSCO, M.; DANTAS, S. O.; COLUCI, V. R.. Simulação de Dinâmica Molecular Usando d Potencial de Lennard-Jones. XII Encontro de Modelagem Computacional, Rio de Janeiro, 2009.
TRAVIESO, G.. Estudo de Processamento Paralelo para Dinâmica Molecular. Universidade de São Paulo, São Carlos, 1989.
