ÁREA
Química Computacional
Autores
Viana, K.P. (UEMA) ; Sousa, A.S. (UEMA) ; Santos, P.L.L. (UEMA) ; Almeida, P.H.B. (UEMA) ; Fernandes, R.M.T. (UEMA) ; Khan, A. (UEMA)
RESUMO
Este trabalho apresenta os cálculos de otimização e frequência realizados na molécula de acetato de metila e seus radicais nas temperaturas 0K, 298K, 500K, 1000K e 1500K pelo método DFT na base Pople. O acetato de metila, possui baixa massa molecular, temperatura e pressão estáveis em condições ambientes tornou-se um candidato à biocombustível por mostrar-se, segundo os cálculos realizados, semelhante ao etanol e com algumas propriedades termodinâmicas com resultados teoricamente mais favorável, o que faz com que esse éster apresente produtos de combustão pouco nocivos. Os cálculos visam realizar uma avaliação termoquímica e cinética da decomposição térmica das matérias como combustíveis alternativos em busca de compreender as propriedades termoquímicas e mecanismo da reação elementar.
Palavras Chaves
Acetato de metila; Termodinâmica; Combustível alternativo
Introdução
Desde o século XX o petróleo e seus derivados foram as principais fonte de combustível do mundo. Porém, como toda fonte não renovável, o medo do esgotamento das reservas petrolíferas tornou-se presente na sociedade contemporânea, o que motivou estudos sobre novas fontes de combustíveis. Atualmente existem os biocombustíveis como o etanol, os provenientes do acetato de metila e etila, entre outros. O desenvolvimento desses combustíveis alternativos deixou de ser pela preocupação com o esgotamento das reservas e passou a ser pela pressão dos ambientalistas sobre as indústrias automobilísticas “A indústria automotiva tem modernizado continuamente a tecnologia dos motores, buscando uma queima mais eficiente dos combustíveis e uma redução da emissão de gases, visando atender às crescentes exigências ambientais” (GOLDENSTEIN e AZEVEDO, p.244, 2006). Os avanços na Química Computacional foram os principais responsáveis pelos estudos teóricos desses biocombustíveis, uma vez que permitiu a experimentação em meio laboratorial com resultados rápidos e sem grandes custos. O entendimento dos estudos termodinâmicos, que estudam as interações da energia nas quais as variações de temperaturas são importantes também impulsionaram a química computacional e o desenvolvimento de experiências teóricas, pois o comportamento dos combustíveis em diversas temperaturas é algo crucial a se conhecer antes do uso comercial, uma vez que negligenciado, pode interferir no seu bom desempenho e colocar vidas em risco, como por exemplo, o acetato de metila, assim como outros combustíveis com etanol, gasolina e diesel são altamente inflamáveis (PEREIRA e BAPTISTA, 2022) O estudo termodinâmico e cinético das reações do acetato de metila (também chamado de etanoato de metila) provém da necessidade do controle desse composto, pois o mesmo apresenta subprodutos, que podem causar problemas para a indústria, o que tornou o estudo viável a partir da complexidade em se propor um modelo cinético de combustão a partir de parâmetros cinéticos para moléculas de ésteres de massa molecular elevada. Muitos estudos ainda serão realizados sobre as reações desse composto, como a cinética da abstração do hidrogênio deles, pois estão associados ao processo de ignição do combustível, por ora, este trabalho visa apenas realizar uma avaliação termoquímica e cinética da decomposição térmica das matérias como combustíveis alternativos em busca de compreender as propriedades termoquímicas e mecanismo da reação elementar. Para (MARQUES e BOTTI, 2006) o DFT surgiu como uma forma de facilitar e melhorar o cálculo de moléculas, oferecendo resultados mais rápidos e precisos por meio da química computacional. O cálculo de grandes moléculas foi o mais beneficiado com o desenvolvimento deste método, o que contribuiu para o avanço da Química Quântica nas demais pois contribuiu para a invenção de novos matérias, e tendo aplicações importantes no estudo de metais e semicondutores. Com a ajuda de computadores a DFT permitiu estudar sistemas cada vez mais complexos, o que influenciou no estudo de sólidos, moléculas e átomos, além de ter um papel fundamental nas áreas nanotecnologia e biotecnologia. Observados todas essas possibilidades graças ao DFT, o comitê do Nobel de Química atribuiu o prêmio á Walter Kohn e John Pople. Apesar disto, a introdução desta teoria na Química foi lenta. De acordo (MARQUES e BOTTI, 2006) essa introdução apenas tornou-se possível com a contribuição de J. Pople que a introduziu no seu programa de computador Gaussian, tornando-se o programa de Química Quântica mais usado em todo o mundo. A Energia Livre de Gibbs (G), em termos básicos, é a energia necessária de que um processo necessita para realizar trabalho em temperatura e pressão constantes. Por outro lado, (CHEN, 2019) defende Energia Livre de Gibbs molar, ou Energia livre de GIBBS parcial, em J/mol deve ser identificada com um potencial químico, uma vez que o potencial químico de uma substância é a energia química por mol da substância, o que deste modo, a Energia Livre de Gibbs é a energia química e a substância pode ser pura ou um sistema multicomponente. Por outro lado, a Entalpia (H) é a energia térmica que cada composto presente em uma reação possui, ou seja, é a energia interna da molécula. A entalpia também pode ser entendida como a energia, sobre pressão constante, poderia ser transformada em calor. Já a Energia Interna é quando um sistema é capaz de realizar trabalho, sendo a soma de todas as energias que um sistema possui em seu interior, estando associada a seus átomos e moléculas do ponto de vista de um referencial, estando esse em repouso.
Material e métodos
O trabalho faz uso de métodos baseados na DFT, com a base de Pople e o tempo médio de cada cálculo foi em torno de 5min no Notebook da Lenovo Ideapad S145, 8ª Geração com processador Intel Core i5, 8GB de RAM e 1TB de armazenamento. Sobre os cálculos das moléculas de acetato de metila, primeiro foi calculado sobre a molécula completa, depois alguns átomos iam sendo retirados e os íons iam sendo calculados, como anexado abaixo: Após isto, todos os radicais obtidos a partir dessa composição foram desenhados e calculados no GaussianView e Gaussian 09W, respectivamente. Após o desenho de cada estrutura da molécula, veio a parte do cálculo. No Gaussian há a opção de “Calculate”, que acessada mostra os parâmetros para os cálculos. Os utilizados foram “Otimização e Frequência” Como o processador e RAM do notebook utilizado são rápidos o suficiente para permitir a rapidez dos cálculos, por volta de 4 minutos os resultados já estavam prontos, resultados esses referentes à Entalpia, Energia Livre de Gibbs, Energia Interna, Capacidade Volumétrica e Entropia. Esse mesmo procedimento foi feito 25 vezes para a molécula principal e seus 4 radicais nas cinco temperaturas trabalhadas (0K, 298K, 500K, 1000K e 1500K). Em seguida, a escolha do método, DFT, na base Pople. Ao final de cada cálculo, eles foram extraídos e tabelados no Excel para os devidos cálculos. Com todos os resultados tabelados e transformados para J/mol (Energia de Gibbs (G) e Entalpia(H) estavam em Hartree; Entropia (S), Capacidade Volumétrica (CV), estavam em cal/mol e a Energia Interna (U) estava kcal/mol.Para encontrar o valor da Capacidade Térmica (CP), foi necessário utilizar a seguinte fórmula CP = CV + R, sendo R a contante dos gases em Joule igual 8,31441J/mol. Após encontrados todos os valores, os resultados dos radicais foram reunidos e tabelados. Após essa reunião e comparação dos dados, é possível perceber a baixa variação nos valores entre os próprios radicais e entre estes e o acetato de metila. Na tabela apresentada a seguir, com todos os valores em J/mol na temperatura 0K, é possível perceber uma semelhança nas moléculas parecidas. Os radicas (CH3COOCH2, CH3COO, CH2COOCH3 e COOCH3) a semelhança dos valores entre aqueles que perderam um hidrogênio são parecidos mudando apenas pouco números nas casas decimais. O mesmo ocorre com os radicais que perderam a metila, independente do lado da perda, os resultados se mantêm constantes, com apenas algumas diferenças nas casas decimais. Por último, as moléculas com diferenças nos valores mais acentuadas são aquelas que apresentam número de átomo/ligações mais distantes entre si. E é possível perceber também que essas características se repetem em todas as temperaturas. Todos os cálculos de estrutura eletrônica e parâmetros termodinâmicos foram feitos utilizando o programa GaussView e Gaussian 09W. As temperaturas consideradas foram 0K, 298K, 500K, 1000K e 1500K.
Resultado e discussão
A seguir estão os resultados obtidos através do GaussianView e Gaussian 09W. Ao
observar esses valores, perceber-se à que as variações são bem discretas e essa
baixa variação nos valores é algo muito importante quando a questão é
estabilidade do composto. Ter um composto químico que apesar da mudança de
temperatura não sofre grandes mudanças é algo muito relevante a se estudar, no
caso de combustíveis é essencial, pois demonstra que aquele componente, mesmo em
carros com mais ou menos resistência e queima gasto de energia se manterá
constante, independente da temperatura. A questão do abastecimento em dias frios
e quentes também entra nessa discussão na questão da Capacidade Volumétrica e
Capacidade Térmica dos combustíveis.
Desta forma, tanto na variação de entalpia quanto nas próximas variações,
perceber-se-á que apesar de sofre aumentos nos valores, serão bem discretos, uma
vez que o carbono, o hidrogênio e o oxigênio impedem uma variação muito alta.
Um outro ponto é que a entalpia sofre um grande aumento a partir da faixa de 298
a 500K, uma vez que é nessa faixa que ocorre a autoignição. Também, utilizando a
molécula do etanol como padrão de comparação dos valores termoquímicos, a
entalpia experimental desse biocombustível é na faixa de 20E+4J segundo (ATKINS,
2008; FÉREY,2011), logo, os dados encontrados aqui estão nessa faixa.
Para facilitar a demonstração, CH3COOCH3, molécula principal, CH3COOCH2,
Radical I (Rad I), CH3COO Radical II (Rad II), CH2COOCH3 Radical III (Rad III) e
COOCH3 Radical IV (Rad IV) .
A variação da Energia Livre de Gibbs obteve o resultado esperado, uma vez que
(G) baixas indicam uma melhor eficiência, pois exercem trabalho espontaneamente.
A capacidade térmica, por outro lado, demonstrou aumento com a temperatura, isso
porque depende do tamanho e massa da molécula. Como a molécula principal é
maior, ela tem maior capacidade térmica que seus radicais, ou seja, precisa de
maior quantidade de calor para aumentar a temperatura de sua massa.
Como a entropia mede o grau de desordem de combustão do etanol, ou seja, o
transforma de líquido para gás, o valor padrão dela para o etanol é 1,60E+02
(NIKOLAU e FILHO, 2020) os resultados encontrados na primeira variação são
próximos e quanto maior a variação de temperatura, maior a entropia, ou seja,
maior o grau de desordem.
Com o aumento da temperatura, o aumento da energia interna é justificável pelo
aumento da energia cinética e potencial, já que U é a soma dessas duas, ou seja,
é a quantidade total de energia relacionada ao movimento dos átomos e moléculas
presentes naquele sistema. Todas as variações mantiveram-se constantes, em
relação às moléculas semelhantes. Os únicos valores com uma variação maior são o
CH3COO (1) e o COOCH3 (2), isso devido à perda do grupo metil mais próximo da
dupla ligação no composto.
Em todas as variações, a constância dos valores permanece, apesar de em
comparação à primeira variação (298K-0K), a última variação (1500K-0K) ter
apresentado valores maiores, mas isso é justiçável pelo aumento de energia no
composto. Observados todos os radicais e comparando seus valores com a molécula
principal, todas as variações apresentam resultados maiores, isso também é
justificável pela quebra da molécula. Um radical (cátion), instável no ambiente
gasta mais energia que uma molécula estável, logo a justificativa para os
valores serem mais altos.
Nota-se que o Rad II e o Rad IV independente da temperatura têm um valor muito
próximo de zero, o que pode significar que a taxa da reação em relação à
temperatura é desfavorável para os produtos. Para o Rad I, temos um valor mais
favorável À partir de 500K e que aumenta gradativamente com a temperatura,
seguindo praticamente a mesma taxa de crescimento em relação à Rad III (números
de casas decimais), o que pode indicar que a interferência na estrutura dos
radicais influencia diretamente na taxa de reação.
O Rad I e o Rad III são estruturas que perderam o hidrogênio em sua composição,
especificamente nos carbonos dos extremos da molécula, o que abriu a
possibilidade de um carbocátion, além disso, a molécula possui certa polaridade
por conta da dupla ligação com o oxigênio e a presença de outro oxigênio na
cadeia (um éster), logo, a chance de formação do radical é maior do que o Rad II
e o Rad IV.
Em relação à Rad II e IV são resultados da retirada do componente CH3, que, dada
a estabilidade da ligação, é necessária mais energia para quebrá-la. Logo a
formação desses intermediários é desfavorecida, mostrando que forma como a
molécula se decompõe em uma reação interfere diretamente na taxa de reação dos
produtos.
Com a base B3LYP-D3, a reação de formação do Rad III apresentou valores da
Energia Livre de Gibbs mais baixos que a formação do Rad I em todas as variações
de temperatura, cujos valores comparados com (PEREIRA e BAPTISTA, 2022) são
semelhantes. Enquanto para o Rad I, esse autor encontrou 7,22E4 J/mol e para o
Rad III 6,05E4 J/mol, na variação 298,15K-1K o valor encontrado nesse trabalho
foi respectivamente 4,73E4 J/mol e 4,73E4 J/mol (os valores diferem nas casas
decimais menores) na variação 298K-0K, algo que demonstra uma estabilidade por
parte da molécula, além de apresentarem, do ponto de vista termodinâmico, um
valor consistente nos resultados.
Os valores encontrados de K (Constante da Taxa de Reação em relação à
Temperatura) para cada radical em relação à molécula principal em 5K (Kelvin)
são praticamente iguais a zero, embora não sejam realmente iguais a zero, mas
matematicamente o cálculo da constante nessa temperatura, por estar próxima do
zero absoluto em Kelvin, apresenta resultado positivo, mas com numeração de 10E-
200, é a partir da temperatura 298K que é possível identificar a mudança no
comportamento dos espécimes químicos.
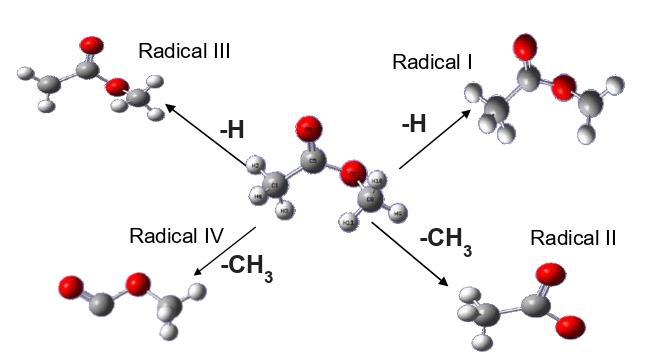
Molécula do Acetato de Metila e seus radicais \r\nintermediários
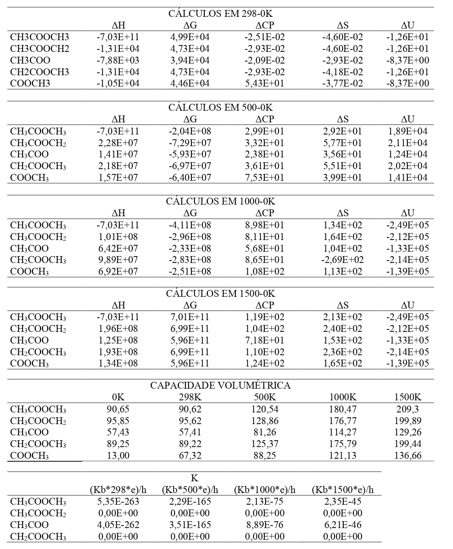
Cálculos obtidos no GaussianView e Gaussian 09W do \r\nAcetato de Metila e seus radicais
Conclusões
Entre tantas fontes alternativas para diminuir a emissão de gases do efeito estufa e ao mesmo tempo manter a eficiência de combustão, o acetato de metila apresenta um comportamento muito favorável por suas propriedades termoquímicas. Sendo um éster que possui estabilidade a temperatura ambiente, sua capacidade de energética na formação de trabalho mostrou-se satisfatória, uma vez que a entropia de seus radicais são estáveis e a variação de energia livre é a favor da espontânea do processo reacionário. Logo, mostra-se tão eficiência quanto os combustíveis que estão no mercado atual, como o próprio etanol (biocombustível mais comum no Brasil) e a gasolina, sendo, portanto, viável sua aplicação no cotidiano seja em motores automobilísticos, seja em outros setores como o aeroespacial. Por se tratar de um teste que por meio de ensaios gastaria muito reagente além das despesas com a produção, equipamentos de analise e etc., o uso da vertente computacional se mostre muito útil uma vez que tem baixo custo econômico e já tem credibilidade atualmente, sendo utilizado com diferentes “métodos” que podem se adequar ao tipo de investigação. Nesse caso, utilizou-se o DFT(B3LYP) pois seus resultados termoquímicos são muito próximos do empírico e utiliza uma porcentagem considerável de um computador, sendo que ele utiliza a densidade eletrônica no lugar da função de onda para determinar propriedades químicas/físicas. Portanto, o Acetato de Metila mostrou-se ser um combustível alternativo, capaz de ter tanta eficiência quanto o etanol, além de ter impacto menor na natureza com a emissão de gases nocivos, podendo ser utilizado para o desenvolvimento de novas tecnologias capazes de utilizar o máximo possível o seu potencial de queima.
Agradecimentos
Agradeço à DEUS pelas bençãos. Agradeço ao Profº. Dr. Alamgir Khan e à UEMA pela oportunidade. Agradeço à minha família, Arthur Sousa, Pedro Santos e Pedro Almeida pela ajuda e companheirismo.
Referências
ATKINS, J.; PAULA, J. Físico-Química, 8ª. Edição. Rio de Janeiro. LTC Editora. 2008.
BIZZO, W. A. EM 722-Geração, Distribuição e Utilização de vapor. Notas de Aula: Capítulo, 2012. Disponível em https://www.fem.unicamp.br/~em672/GERVAP2 .Acessado em 12/08/2023. Acessado em 24/08/2023
BRONDANI, L. N. Produção de biodiesel a partir de matéria-prima com alto teor de acidez: modelagem cinética e termodinâmica, simulação e otimização. 2022. Tese de Doutorado. Universidade Federal de Santa Maria. Disponível em: http://repositorio.ufsm.br/handle/1/27534. Acessado em 17/08/2023.
PEREIRA, S. L.; BAPTISTA, L. Estudo cinético da abstração do hidrogênio do acetato de metila. UERJ, 2022. Disponível em: https://doi.org/10.21826/viiiseedmol202040. Acessado em 18/08/2023.
CHEN, L. Chemical potential and Gibbs free energy. Mrs Bulletin, v. 44, n. 7, p. 520-523, Cambridge, 2019. Disponível em https://doi.org/10.1557/mrs.2019.162. Acessado em 18/08/2023.
GOLDENSTEIN, M.; AZEVEDO, R. L. S. Combustíveis alternativos e inovações no setor automotivo: será o fim da" era do petróleo”? Banco Nacional de Desenvolvimento Econômico e Social, 2006. Disponível em: http://web.bndes.gov.br/bib/jspui/handle/1408/2531. Acessado em 17/08/2023.
FEREY, G.; SERRE, C.; DEVIC, T.; MAURIN, G.; JOBIC, H; LLEWELLYN, P. L.; DE WERELD, G.; VIMONT, A.; DATURI, M.; CHANG, J.S. Why hybrid porous solids capture greenhouse gases? Chemical Society Reviews, v. 40, n. 2, p.550-562, 2011. Disponível em https://doi.org/10.1039/C0CS00040J. Acessado em 23/08/2023.
HAWKEN, Paul. Capitalismo natural. 1 ed. São Paulo: Editora Cultrix, 2000.
MARQUES, M. A.; BOTTI, S. O que é e para que serve a Teoria dos Funcionais da Densidade. Gazeta de física, v. 29, n. 4, p. 10-15, 2006. Disponível em: https://www.tddft.org/bmg/files/papers/5038049.pdf. Acessado em 18/08/2023.
MORGON, N. H.; CUSTODIO, R. Teoria do funcional de densidade. Química Nova, v. 18, n. 1, p. 44-55, 1995. Disponível em: http://submission.quimicanova.sbq.org.br/qn/qnol/1995/vol18n1/v18_n1_10.pdf. Acessado em 18/08/2023.
NIKOLAU, S.; FILHO, A. G. S. O. Química Geral: Entropia e Energia Livre de Gibbs. USP, 2020. Disponível em https://edisciplinas.usp.br/pluginfile.php/5751183/mod_resource/content/1/aula26_entropia_e_gibbs.pdf. Acessado em 10/08/2023.
OCHTERSKI, J. W. Thermochemistry in gaussian. Gaussian Inc, v. 1, p. 1-19, 2000. Disponível em https://www.cup.uni-muenchen.de/ch/compchem/G98thermo.pdf. Acessado em 22/08/2023.
RIZZO, Guilherme Augusto. Simulações de dinâmica molecular aplicadas à combustão de biodiesel. Repositório Digital, 2013. Disponível em: https://www.lume.ufrgs.br/handle/10183/85658. Acessado em 13/07/2023.
SOUZA, A. M. G. P. Termodinâmica Química. CESAD, 2009. Disponível em: https://cesad.ufs.br/ORBI/public/uploadCatalago/19133916022012Termodinamica_Quimica_Aula_1.pdf. Acessado em 22/07/2023.
SOARES, C. V. Simulação de processos de adsorção molecular em material nanoporoso constituído por tereftalato e zircônio. Repositório Digital, 2016. Disponível em: https://repositorio.ufjf.br/jspui/handle/ufjf/1819. Acessado em 22/07/2023.