ÁREA
Química Computacional
Autores
Araujo, M. (UFPI) ; Girão, E. (UFPI) ; Porto, J. (UFPI)
RESUMO
Tripentafenos(TPH) são células construídas através de uma organização 2D concebidas de maneira teórica através de acoplamento de moléculas base de acenpentaleno(ACP). Nesse trabalho foi realizado um estudo teórico baseado no método de funcional de densidade(DFT) para investigar o comportamento eletrônico de 3 diferentes alotropos do ACP nomeados como estruturas alfa, beta e sigma. Essa metodologia conseguiu apresentar de maneira satisfatória a distribuição espacial desses modelos, suas transições mais importantes, mas foi na estrutura de ressonância onde foi determinado as propriedades eletrônicas os estados deslocalizados de cada sistema e assim poder observar um comportamento metálico em apenas um dos modelos enquanto os demais comportam-se como semicondutores
Palavras Chaves
DFT; Tripentafeno; Densidade dos estados
Introdução
As nanoestruturas de carbono atualmente são consideras como potenciais candidatas para pesquisas envolvendo a substituição do silicone em futuras aplicações em nanoestruturas. Essas estruturas tem uma ampla variedade de arranjo espacial, tais como, organização em 0D, 1D, 2D e 3D, além dessa ampla possibilidade de arranjo espacial esses compostos possuem propriedades ajustáveis, fácil síntese e uma enorme facilidade de integração com dispositivos eletrônicos(SILVA et al., 2020). Os mais recentes estudos na área estão focados em modificações físicas e químicas no grafeno com o objetivo de direcionar esses produtos modificados para utilização em dispositivos eletrônicos, sendo que todas essas alterações são realizadas em escala nano. Originalmente as ideias de modificação nas estruturas do carbono para criação de novos alótropos é motivado pela capacidade de rearranjo dos átomos de carbono em formar cadeias baseados nas suas hibridizações, sendo essas combinações podendo ser puras ou um mix entre sp2 e sp3 (ZHAO et al., 2019) , formando assim células unitárias compostos por rearranjos de blocos moleculares (LU; LI, 2013). Essa construção em forma de blocos moléculas se assemelha bastante as rotas de sínteses conhecidas como bottom-up, e com isso o material hipotético criado a partir do alongamento de cadeias de carbono sp² se assemelha mais a uma possível construção experimental, sendo assim, a caracterização in sílico de propriedades eletrônicas como a capacidade semicondutora do material vai estar bem próxima do resultado real.(TERRONES et al., 2000) Nesse trabalho foram propostas 3 diferentes estruturas que são alótropos teóricos 2D de cadeias carbônicas baseadas na estrutura base que é o Tripentafeno essa estrutura foi linkada baseado em diferentes arranjos
Material e métodos
Os cálculos das estruturas eletrônicas foram realizados usando a teoria do funcional da densidade (DFT)- Density Functional Theory, método esse implementado no pacote de códigos do SIESTA(HEYD; SCUSERIA; ERNZERHOF, 2003), os elétrons do caroço ou os elétrons da camada mais interna dos átomos foram descritos através do pseudo potencial de Troullier-Martins(TROULLIER; MARTINS, 1991), as funções de onda foram expandidas usando funções duplamente polarizadas (DZP), o funcional de correlação de troca foi descrito pelo gradiente de aproximação generalizada (GGA), e parametrizado pelo Perdew-Burke-Ernzerhof(PBE) (PERDEW; BURKE; ERNZERHOF, 1996), para as zonas de Brillouin o pacote de Monkhorst foi o escolhido pois acomoda melhor sistemas com células primitivas maiores, a relaxação estrutural adotou uma tolerância de força máxima de 10-2 e V e cada átomo teve uma tolerância de 0.1 GPa.
Resultado e discussão
O objetivo do trabalho era descrever as propriedades eletrônicas dos compostos
sintetizados e observar seu comportamento, e isso foi possível através da
análise dos dados da DOS (densidade dos estados) e da estrutura eletrônica de
bandas, esses dados foram compilados para cada um dos sistemas estudados, sendo
o primeiro a ser analisado é o do TPH-α( Figura 1),nesse caso pode-se observar,
que o sistema tem um comportamento metálico, pois duas bandas, cruzam o nível de
Fermi em torno do ponto M , nesse caso especifico, tanto a banda condução,
quanto a banda de valência cruza o nível de Fermi , sendo assim seus estados
semi preenchidos e assim, qualquer nível de energia é capaz de causar uma
excitação eletrônica, na figura 2 observa-se a distribuição das bandas das
outras formas do alótropos estudados, nesses casos, observa-se que nenhuma das
bandas cruzam o nível de Fermi, e com isso, elas se comportam como
semicondutores com um baixo Gap de transição eletrônica, entre os dois casos do
TPH-β e do TPH-σ, o primeiro caso observa-se um Gap de energia mais baixo, com
mais pontos de singularidade de van Hove, que reforça a capacidade de condução
do material com maior contribuição das bandas deslocalizadas
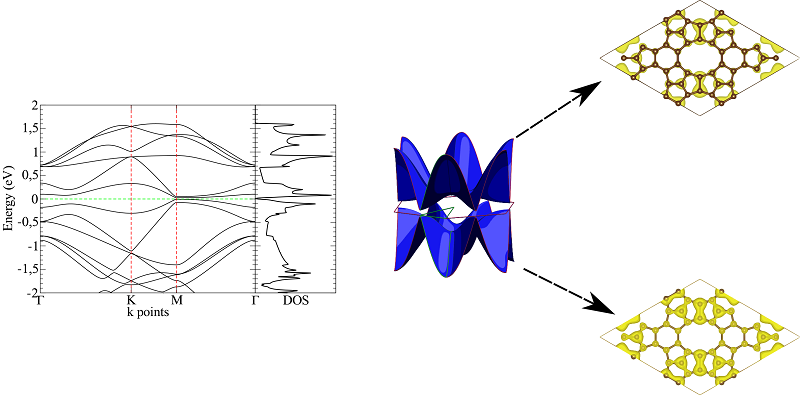
Estrutura de bandas, DOS e LDOS, e bandas plotadas \r\n3D com os respectivos mapas de superficie
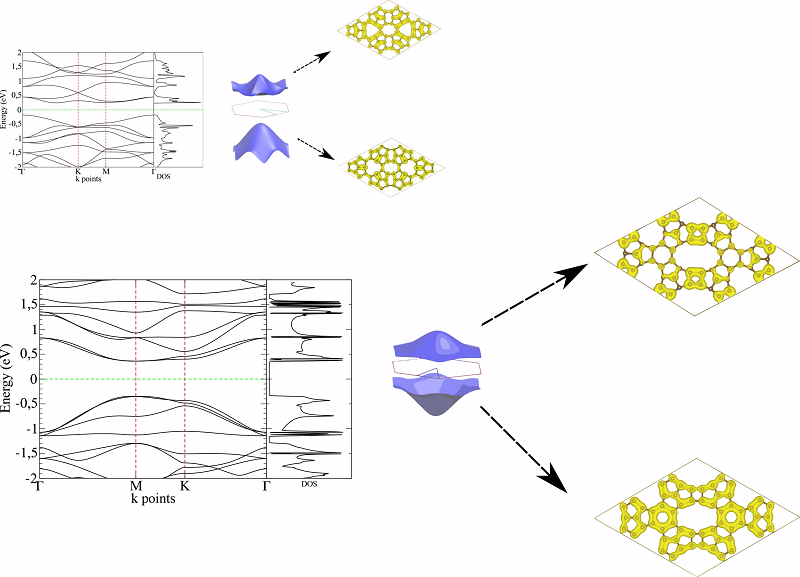
Estruturas de bandas plotadas e mapas de superfície \r\ndos alótropos semicondutores
Conclusões
Em resumo, foi proposto estudar e caracterizar as propriedades eletrônicas de 3 modelos bidimensional de alótropos de carbonos derivados de uma estrutura base, esses alótropos foram chamados de tripentafenos, e caracterizados de acordo com a sua distribuição espacial, sendo eles os modelos alfa, beta e sigma, para cada um desses modelos, pode-se concluir que a estrutura alfa tem um comportamento completamente metálico, enquanto os demais alótropos tem um comportamento de semicondutor, cálculos futuros serão feitos para reforçar os dados com essa propriedade.
Agradecimentos
Referências
HEYD, J.; SCUSERIA, G. E.; ERNZERHOF, M. Hybrid functionals based on a screened Coulomb potential. Journal of Chemical Physics, v. 118, n. 18, p. 8207–8215, 8 maio 2003.
LU, H.; LI, S. D. Two-dimensional carbon allotropes from graphene to graphyne. Journal of Materials Chemistry C, v. 1, n. 23, p. 3677–3680, 21 jun. 2013.
PERDEW, J. P.; BURKE, K.; ERNZERHOF, M. Generalized Gradient Approximation Made Simple. [s.l: s.n.].
SILVA, P. V. et al. Tripentaphenes: Two-dimensional acepentalene-based nanocarbon allotropes. Physical Chemistry Chemical Physics, v. 22, n. 40, p. 23195–23206, 28 out. 2020.
TERRONES, H. et al. New Metallic Allotropes of Planar and Tubular Carbon. [s.l: s.n.].
TROULLIER, N.; MARTINS, J. L. EfFicient pseudopotentials for plane-wave calculations-IIPHYSICAL REVIEW. [s.l: s.n.].
ZHAO, C. X. et al. C-57 carbon: A two-dimensional metallic carbon allotrope with pentagonal and heptagonal rings. Computational Materials Science, v. 160, p. 115–119, 1 abr. 2019.