ÁREA
Química Computacional
Autores
Silva, A.J.F.W.H.S. (UNIVERSIDADE FEDERAL DA PARAÍBA) ; Ventura, E. (UNIVERSIDADE FEDERAL DA PARAÍBA) ; do Monte, S.A. (UNIVERSIDADE FEDERAL DA PARAÍBA)
RESUMO
Este trabalho corresponde à primeira investigação teórica de dinâmica não adiabática do CH2FCl, para isto utilizou-se TD-DFT (M06-2X)/d-aug-cc-pVDZ (C)/aug-cc-pVDZ (F, Cl, H) em conjunto com o método Surface Hopping. As condições iniciais, o espectro de fotoabsorção UV e as trajetórias semiclássicas foram gerados e calculados usando os programas Newton-X/Gaussian 09. A análise dos espectros demonstrou deslocamentos das bandas teóricas n-σCCl* (0,14 eV) e n-3s (0,206 eV) em relação às correspondentes bandas experimentais. Os rendimentos dos fotoprodutos foram de 87% (Cl), 9% (H), 3% (HCl) e 1% (F). Ademais, 18% das simulações de dinâmica seguiram o canal de formação do par iônico, H2CF+ (1Aʹ)/Cl- (1S).
Palavras Chaves
HCFC-31; Fotoquímica computacional; Dinâmica não-adiabática
Introdução
Muitos compostos interferem diretamente no equilíbrio químico O2 ⇌ O3 presente na Estratosfera. Uma das classes de moléculas envolvidas nesse processo são os hidro-cloro-fluorocarbonetos (HCFCs), um exemplo é o CH2FCl (ou HCFC-31). Por apresentarem pelo menos uma ligação C–H, essas substâncias são mais suscetíveis a reações de oxidação com a hidroxila na Troposfera e, portanto, espera-se que uma fração menor dessas substâncias atinja a Estratosfera (ESSA; MOHAMED, 2020; NEWNHAM; BALLARD, 1995). Além disso, estas moléculas possuem átomos de cloro e podem reagir com um mecanismo semelhante aos CFCs, e reduz a concentração de ozônio atmosférico a partir da liberação do radical Cl• após a absorção de radiação UV (ABAS et al., 2018). O estudo do mecanismo da fotodissociação dos CFCs e HCFCs é fundamental para entender como essas moléculas são ativadas na Estratosfera. No caso dos HCFCs a fotoquímica é considerada bastante complexa, e o caminho de dissociação pode levar a diferentes produtos, tanto pela quebra de ligações C–Cl, C–H ou C–F (gerando como produtos Cl atômico, HF, HCl ou FCl), como também pela quebra da ligação C–C (RODRIGUES et al., 2019). A fim de obter uma compreensão teórica completa dos mecanismos fotoquímicos, é necessário determinar todas as importantes vias não-adiabáticas e adiabáticas disponíveis após a fotoexcitação. O objetivo desta pesquisa é investigar os mecanismos de fotodissociação do 1-cloro-1-flúor-metano (CH2FCl), utilizando um método de estrutura eletrônica single reference (TD-DFT) e a dinâmica não-adiabática através do método Surface Hopping.
Material e métodos
Etapa 1) Os programas Newton-X (BARBATTI et al., 2022) em interface com o Gaussian 09 (FRISCH et al., 2016) aliado ao método TD-DFT (M06-2X) com a função de base d-aug-cc-pVDZ (C)/aug-cc-pVDZ (H, F, Cl) foram utilizados para o cálculo das condições iniciais de dinâmica e geração dos espectros de fotoabsorção para o CH2FCl na região do UV. Uma geometria de mínimo previamente otimizada com o método e base citados foi usada como input. Além disso, foram incluídos os cinco primeiros estados singletos. Estudos das transições na região de Franck-Condon mostram que esse conjunto de estados é suficiente para atingir o estado iônico (estrutura de mínimo na superfície S3). O espectro foi calculado utilizando a abordagem da seleção de um conjunto de posições nucleares (do inglês nuclear ensemble approach), que apesar de fornecer as intensidades absolutas e larguras de banda, não inclui resolução vibracional (CRESPO-OTERO; BARBATTI, 2012). Esses espectros foram obtidos a partir de uma amostragem de 1000 pontos, utilizando a distribuição de Wigner (LEYVA et al., 2011). Etapa 2) As simulações de dinâmica não-adiabática no estado gasoso foram calculadas na janela de excitação 7,8 ± 0,25 eV (154-164,2 nm), os quais 100 trajetórias foram escolhidas estocasticamente e, posteriormente calculadas. As equações de movimento clássicas foram integradas a cada 0,5 fs. Utilizaram-se os mesmos softwares, métodos, base e quantidade de estados da etapa anterior.
Resultado e discussão
Experimentalmente, a fotoquímica do HCFC-31 foi estudada por Doucet et al. (1973) a partir da espectroscopia de absorção UV de vácuo em comprimentos de onda de 120 a 200 nm (10,3 a 6,2 eV). O espectro teórico de absorção UV obtido para o CH2FCl foi calculado a nível TD-DFT com o funcional M06-2X, com a base d-aug-cc-pVDZ (C)/aug-cc-pVDZ (H, F, Cl). Um estudo anterior de benchmarking apontou o M06-2X como o funcional que melhor descreveu as energias de excitação vertical, as configurações eletrônicas e a geometria de mínimo do par iônico. Ambos os espectros se encontram na Figura 1. No espectro experimental estão presentes três bandas: I) a de mais baixa intensidade, centrada em 7,75 eV; II) outra com maior intensidade entre 8,0 e 8,5 eV com picos distintos, respectivamente, em 8,10 e 8,38 eV; e III) a de intensidade mais alta entre 8,6 e 9,6 eV, todavia com um ombro em 8,82 eV e quatro picos diferenciados em 9,02, 9,28, 9,44 e 9,59 eV. Nota-se que as bandas espectrais de energia mais baixa de cada metodologia (TD-M06-2X e experimental) apresentam uma boa concordância (por volta de 7,8 eV), porém com diferentes valores de σ (seção de choque de fotoabsorção). Isso demonstra que há transições eletrônicas com valores relevantes de forças do oscilador. Ademais, o perfil da segunda banda experimental (8,1-8,4 eV) é muito parecido com o perfil da segunda banda do espectro TD-DFT (8,1-8,5 eV). Doucet e colaboradores (1973) assinalaram as bandas como referentes a transições n-σCCl* (I), n-3s (II) e n-3p (III), enquanto o espectro teórico descreveu apenas estados n-σCCl* e n-3s. Onde n corresponde aos orbitais não ligantes do cloro e 3s e 3p são orbitais de Rydberg do carbono. Observa-se um grande deslocamento das bandas teóricas n-σCCl* (0,14 eV) e n-3s (0,206 eV) em relação às correspondentes bandas experimentais na Figura 1. Por conseguinte, a quantidade de estados utilizada (4 estados excitados) para a criação do espectro teórico não é suficiente para a descrição das bandas experimentais n-3p. Diversos trabalhos afirmam que quanto maior a fração de troca exata (fator HF) presentes nos funcionais híbridos, maior o erro médio positivo das energias de excitação, ou seja, os funcionais estudados superestimaram a energia média experimental. Esta tendência é bastante geral para estados excitados de baixa altitude (FERRÉ et al., 2016; DIERKSEN; GRIMME, 2004). Desta forma, pode-se afirmar que o M06-2X (fator HF 54%) superestima o gap de energia entre o estado fundamental e o excitado, o que reflete sua absorção em comprimentos de onda menores (energias maiores) do que os experimentalmente observados, o que é uma característica comum desse tipo de funcional. Recentemente, estudos computacionais de haletos de alquila, incluindo CFCs e HCFCs, utilizando os métodos altamente correlacionados CASSCF e MR-CISD(+Q) com bases extensas demonstraram a presença de um estado excitado que leva a formação de fragmentos iônicos, como o cloreto (BEZERRA et al., 2021; MEDEIROS et al., 2018; RODRIGUES et al., 2016; RODRIGUES et al., 2019; SILVA, 2019; VENTURA; DO MONTE, 2020). O mecanismo de liberação de Cl− inicia-se por meio de excitações de alta energia para estados de Rydberg, por conseguinte, o sistema relaxa rapidamente ao estirar a ligação C–Cl, e sofre uma série de eventos não-adiabáticos (associados a distorções estruturais apropriadas), levando a molécula a estados excitados inferiores, e eventualmente populando o estado de par iônico e produzindo Cl−. Das 100 trajetórias inicialmente consideradas, 18% finalizaram no estado S3. Isto significa que essa porcentagem de trajetórias segue o canal de formação do par iônico, H2CF+ (1Aʹ) / Cl- (1S), e leva a liberação de Cl-. Esse valor é considerado alto e relevante quando comparado com dados de dinâmica não-adiabática do CF3CH2Cl (HCFC-133a), a nível TD-ωB97XD com a base d-aug-cc-pVDZ (Cl)/aug-cc-pVDZ (C, F, H) cujo rendimento de cloreto foi de 15% partindo de uma excitação de 11,2 ± 0,25 eV (RODRIGUES et al., 2019). O alto valor de rendimento quântico de cloreto numa janela de excitação mais baixa (7,8 ± 0,25 eV) é considerado um importante resultado levando em consideração a importância da liberação de Cl- na Estratosfera para o equilíbrio O2 ⇌ O3, uma vez que esse íon não interfere nesse equilíbrio. A Figura 2 mostra uma trajetória representativa para a dinâmica do CH2FCl. Pode-se observar que, no início da simulação os quatro estados excitados possuem energias muito próximas, mais precisamente S1/S2 e S3/S4 cujas energias são 6,7/7,0 eV e 8,0/8,4, respectivamente. Em 4 fs, ocorre o “salto” do S3 para o S4, e aos 14,5 fs o sistema retorna para S3. Outro salto acontece em 37 fs, o qual o sistema vai de S3 para S1, no entanto, após 1 fs o sistema retorna para S3. Nota-se também que dos 62,5 aos 91 fs o sistema permanece em S2, onde provavelmente ocorre uma interseção cônica entre o segundo e o terceiro estados excitados. Nos primeiros fentossegundos algumas distorções na molécula são observadas, por exemplo, pequenas variações em todas as ligações e ângulos de ligação. Próximo a 90 fs o comprimento de ligação C−Cl começa a aumentar e o cloro tende a se dissociar do resto da molécula. O aumento no comprimento dessa ligação é acompanhado pela tendência da planarização do grupo CH2F. O rendimento dos produtos encontrados nas simulações a nível TD-DFT foram: 87% (Cl), 9% (H), 3% (HCl) e 1% (F). Nota-se que o produto majoritário, o cloro atômico, possui rendimento quase 10 vezes maior que o segundo produto, o H atômico. Isto significa que a fragmentação do CH2FCl ocorre preferencialmente no estado excitado e que a molécula segue a maioria dos canais de dissociação da ligação C−Cl. Importante frisar que parte da fotólise C−Cl ocorre heteroliticamente, no qual o fotoproduto é o cloreto. A liberação de H atômico pode estar associada a recombinações dos estados excitados com o fundamental, além disso, a ligação C−H tem caráter mais covalente que as ligações C−Cl e C−F e, por conseguinte, se torna mais difícil de ser quebrada. Já a saída do fragmento HCl, embora em menor proporção, pode contribuir para a liberação de Cl e H atômicos. Uma única trajetória levou a liberação de F atômico, no qual esse caminho de dissociação está relacionado com as transições envolvendo o orbital σCF*, apesar dessas transições não estarem presentes nos estados obtidos a nível MR-CISD(+Q) e base d-aug-cc-pVDZ (C)/aug-cc-pVDZ (Cl, F, H) (ver SILVA, 2019). Os resultados da dinâmica estão diretamente ligados às faixas de excitações, e cada uma delas conduz a diferentes canais fotoquímicos. As excitações do estado fundamental com energias mais altas que 8,0 eV levam a estados que são populados prioritariamente por estados do tipo σCCl*, o que resulta principalmente na quebra da ligação C−Cl. Por outro lado, o aumento da população dos estados de Rydberg n-3s e n-3p implica na formação de produtos multifragmentados, destacando a importância desses estados, particularmente os de mais baixas energias, na fotoquímica deste grupo de compostos, os HCFCs (RODRIGUES et al., 2016; RODRIGUES et al., 2019). Excitações ainda mais altas, por volta de 11,49 eV (108 nm) podem levar ao aumento da formação de fragmentos envolvendo o F, pois viu-se que os estados mais energéticos envolvem orbitais não ligantes do F, ainda que os estados mais energéticos sejam altamente degenerados e envolvam muitos estados de Rydberg, como visto nas curvas de energia potencial obtidas a nível MR-CISD(+Q) (SILVA, 2019).
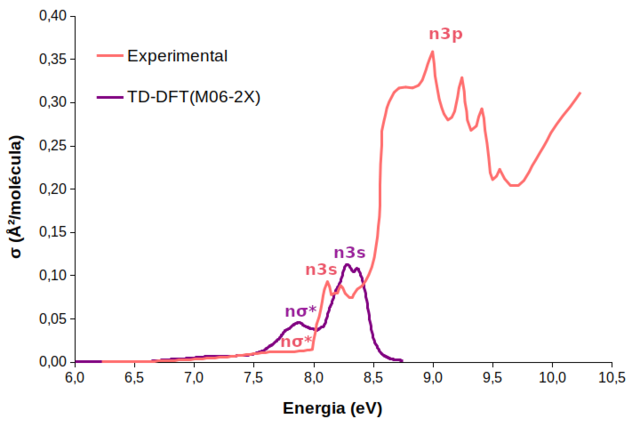
Espectros de absorção UV para o HCFC-31. O espectro experimental foi obtido por Doucet e colaboradores (1973).
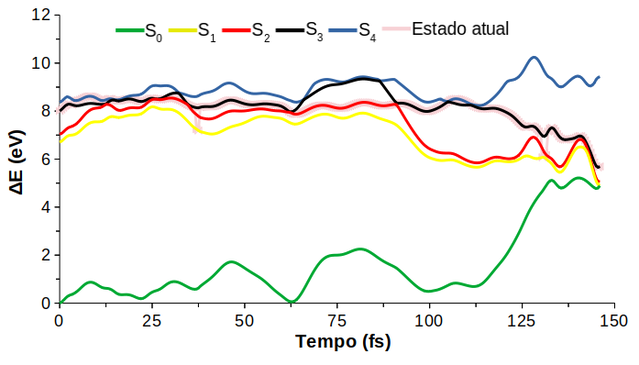
Evolução temporal das energias de uma trajetória representativa com fim em S3 para o HCFC-31 obtida a nível TD-M06-2X.
Conclusões
Este estudo apresenta a primeira descrição teórica a respeito da fotoquímica do CH2FCl, com destaque para a dinâmica não-adiabática utilizando o método de estrutura eletrônica TDDFT aliado ao método Surface Hopping. Houve deslocamentos para energias maiores das bandas n-σCCl* e n-3s do espectro teórico de absorção UV em relação às bandas experimentais. As simulações de dinâmica, situadas na janela de excitação de 7,8 ± 0,25 eV, indicaram um alto rendimento do estado S3, o qual segue o canal de formação do par iônico, HFC+H•••Cl-. Além disso, a maioria das trajetórias tiveram o cloro como fotoprodudo, onde parte da fotólise C−Cl ocorre heteroliticamente, contribuindo para a liberação de Cl- na atmosfera.
Agradecimentos
Ao CNPq, CAPES, FAPESQ, PPGQ-UFPB e, em especial à todos que os cidadãos que lutam pela melhoria da Educação e contra o fascismo e negacionismo científico.
Referências
ABAS, N.; KALAIR, A. R.; KHAN, N.; HAIDER, A.; SALEEM, Z.; SALEEM, M. S. Natural and synthetic refrigerants, global warming: A review. Renewable and Sustainable Energy Reviews, v. 90, p. 557-569, 2018.
BARBATTI, M.; BONDANZA, M.; CRESPO-OTERO, R.; DEMOULIN, B.; DRAL, P. O.; GRANUCCI, G.; KOSSOSKI, F.; LISCHKA, H.; MENNUCCI, B.; MUKHERJEE, S.; PEDERZOLI, M.; PERSICO, M.; PINHEIRO JUNIOR, M.; PITTNER, J.; PLASSER, F.; GIL, E. S.; STOJANOVIC, L. Newton‐X Platform: New Software Developments for Surface Hopping and Nuclear Ensembles. Journal of Chemical Theory Computation, v. 18, p. 6851−6865, 2022.
BEZERRA, M. G.; LEITÃO, E. F. V.; ANDRADE, R. B.; VENTURA, E.; ANDRADE, DO MONTE, S. A. Photochemistry of Monohydrated Chloromethane: Formation of Free and Hydrated Cl- and CH3+ Ions from a Solvent-Shared Semi-Ion-Pair. The Journal of Physical Chemistry A, v. 125, p. 8603−8614, 2021.
CRESPO-OTERO R; BARBATTI M. Spectrum simulation and decomposition with nuclear ensemble: formal derivation and application to benzene, furan and 2-phenylfuran. Theoretical Chemistry Accounts, v. 131, p. 1-14, 2012.
DIERKSEN, M.; GRIMME, S. The Vibronic Structure of Electronic Absorption Spectra of Large Molecules: A Time-Dependent Density Functional Study on the Influence of “Exact” Hartree-Fock Exchange. The Journal of Physical Chemistry A, v. 108, p. 10225-10237, 2004.
DOUCET, J.; SAUVAGEAU, P.; SANDORFY, C. The Journal of Chemical Physics, v. 58, n. 9, 1973.
ESSA, A. G. A.; MOHAMED, A. A. Selection of optimal fluid for refrigeration cycles. World Journal of Advanced Engineering Technology and Sciences, v. 01, p. 21–36, 2020.
FERRÉ, N.; FILATOV, M.; HUIX-ROTLLANT, M. Density-Functional Methods for Excited States, Springer: [s.l.], 2016.
FRISCH, M. J.; TRUCKS, G. W. ; SCHLEGEL, H. B.; SCUSERIA, G. E.; ROBB, M. A.; CHEESEMAN, J. R.;SCALMANI, G.; BARONE, V.; PETERSSON, G. A.; NAKATSUJI, H.; LI, X.; CARICATO, M.; MARENICH, A.; BLOINO, J.; JANESKO, B. G.; GOMPERTS, R.; MENNUCCI, B.; HRATCHIAN, H. P.; ORTIZ, J. V.; IZMAYLOV, A. F.; SONNENBERG, J. L.; WILLIAMS-YOUNG, D.; DING, F.; LIPPARINI, F.; EGIDI, F.; GOINGS, J.; PENG, B.; PETRONE, A.; HENDERSON, T.; RANASINGHE, D.; ZAKRZEWSKI, V. G.; GAO, J.; REGA, N.; ZHENG, G.; LIANG, W.; HADA, M.; EHARA, M.; TOYOTA, K.; FUKUDA, R.; HASEGAWA, J.; ISHIDA, M.; NAKAJIMA, T.; HONDA, Y.; KITAO, O.; NAKAI, H.; VREVEN, T.; THROSSELL, K.; MONTGOMERY JR., J. A.; PERALTA, J. E.; OGLIARO, F.; BEARPARK, M.; HEYD, J. J.; BROTHERS, E.; KUDIN, K. N.; STAROVEROV, V. N.; KEITH, T.; KOBAYASHI, R.; NORMAND, J.; RAGHAVACHARI, K.; RENDELL, A.; BURANT, J. C.; IYENGAR, S. S.; TOMASI, J.; COSSI, M.; MILLAM, J. M.; KLENE, M.; ADAMO, C.; CAMMI, R.; OCHTERSKI, J. W.; MARTIN, R. L.; MOROKUMA, K.; FARKAS, O.; FORESMAN, J. B.; FOX, D. J., Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2016.
LEYVA, V.; CORRAL, I.; FEIXAS, F.; MIGANI, A.; BLANCAFORT, L.; GONZÁLEZ-VÁZQUEZ, J.; GONZÁLEZ, L. A non-adiabatic quantum-classical dynamics study of the intramolecular excited state hydrogen transfer in ortho-nitrobenzaldehyde. Physical Chemistry Chemical Physics, v. 13, p. 14685–14693, 2011.
MEDEIROS, V. C.; ANDRADE, R. B.; RODRIGUES, G. P.; VENTURA, E.; BAUERFELDT, G. F.; BARBATTI, M.; DO MONTE, S. A. Photochemistry of CF3Cl: Quenching of Charged Fragments Is Caused by Nonadiabatic Effects. Journal of Chemical Theory and Computation, v. 14, p. 4844−4855, 2018.
NEWNHAM, D. J.; BALLARD, J. Fourier transform infrared spectroscopy of HCFC-142b vapour. Journal of Quantitative Spectroscopy & Radiative Transfer, v. 53, n. 5. p. 471-479, 1995.
RODRIGUES, G. P.; VENTURA, E.; DO MONTE, S. A.; BARBATTI, M. UV-Photoexcitation and Ultrafast Dynamics of HCFC-132b (CF2ClCH2Cl). Journal of Computational Chemistry, v. 37, p. 675-683, 2016.
RODRIGUES, G. P.; LIMA, T. M. L.; ANDRADE, R. B.; VENTURA, E.; DO MONTE, S. A.; BARBATTI, M. Photoinduced Formation H-Bonded Ion Par in HCFC-133a. The Journal of Physical Chemistry A, v. 123, p. 1953-1961, 2019.
SILVA, ALBERT JORGIVAN FILHO WESLEY HESS DE SOUSA SILVA. Estudo Teórico da Fotoquímica do 1-cloro-1-flúor-metano (HCFC-31). 2019. 77 f. Dissertação (Mestrado em Química) – Departamento de Química, Universidade Federal da Paraíba, João Pessoa, 2019.
VENTURA, E.; DO MONTE, S. A. Hydrogen‐bonded contact ion pair in gaseous chloroethane: a multi‐reference configuration interaction with singles and doubles (MR‐CISD) study including extensivity corrections, Theoretical Chemistry Accounts, v. 139, 2020.