ÁREA
Química Computacional
Autores
Silva, A.J.F.W.H.S. (UNIVERSIDADE FEDERAL DA PARAÍBA) ; Ventura, E. (UNIVERSIDADE FEDERAL DA PARAÍBA) ; do Monte, S.A. (UNIVERSIDADE FEDERAL DA PARAÍBA)
RESUMO
Neste estudo, avaliou-se o desempenho de sete funcionais TD-DFT na reprodução de certas propriedades fotoquímicas do CH2FCl. A comparação foi feita com dados MR-CISD(+Q). Descobriu-se que M06-2X é um dos funcionais mais exatos para avaliar energias de excitação eletrônica. Além disso, o BHandHLYP também apresentou boa concordância com os dados do médoto multireferência, porém este funcional não reproduziu os estados σCCl-3pσ e σCCl-σCCl*. Houve uma boa similaridade nos parâmetros geométricos da estrutura do estado iônico dos funcionais BHandHLYP, M06 e M06- 2X com a estrutura de referência. Contudo, o M06-2X teve a melhor performance com relação às energias de excitação vertical e na natureza dos estados com relação ao MR-CISD+Q.
Palavras Chaves
CH[sub]2[/sub]FCl; Estados excitados; Fotoquímica computacional
Introdução
O trabalho pioneiro de Molina e Rowland (1974), representou um marco no entendimento da química da atmosfera. Foi sugerido que substâncias que eram amplamente utilizadas pela indústria, os cloro-fluorocarbonetos (CFCs), não eram removidos do ambiente por mecanismos de limpeza comuns que operam na baixa atmosfera. Logo, a estabilidade dessas substâncias poderia permitir que a maior parte delas alcançassem a Estratosfera, onde ocorre absorção de radiação ultravioleta e dissociação, com liberação de cloro atômico. O cloro, liberado fotoquimicamente a partir dos CFCs, interfere no ciclo natural do ozônio transformando O3 em O2 e, portanto, reduzindo a quantidade de ozônio disponível. A comprovação da redução do ozônio estratosférico só ocorreu em 1985 (SHANKLIN, 2010). Essas substâncias foram substituídas pelos hidro-cloro-fluorocarbonetos (HCFCs), que por apresentarem pelo menos uma ligação C–H, são mais suscetíveis a reações de oxidação com a hidroxila na Troposfera e, portanto, espera-se que uma fração menor dessas substâncias atinja a Estratosfera (ESSA; MOHAMED, 2020). Estas moléculas possuem átomos de cloro e podem reagir com um mecanismo semelhante aos CFCs (ABAS et al., 2018). O HCFC-31 ou Freon-31 ou R-31 (refrigerant-31) são os nomes industriais da molécula 1- cloro-1-flúor- metano, CH2FCl. As preocupações crescentes sobre os efeitos ambientais adversos dos hidrocarbonetos halogenados têm motivado as inúmeras investigações experimentais e computacionais realizadas nas últimas décadas visando sua caracterização. Por se tratar de um HCFC, sua liberação na atmosfera catalisa o consumo de moléculas de ozônio e amplia os efeitos do aquecimento global devido aos estiramentos das ligações C–F e C–Cl absorverem energia na faixa do infravermelho (8 a 12 μm). Contudo, o HCFC-31 possui a vantagem de poder ser removido da atmosfera pela reação com os radicais OH•, levando à formação principalmente de H2O e HCFCl• (BHATNAGAR; CARR, 1996; CHARMET et al., 2013). Estudos de referência, ou simplesmente benchmarking, que investigam o desempenho de métodos químicos quânticos para o cálculo de estados eletronicamente excitados de sistemas moleculares geralmente se concentram nas energias de excitação (LOOS et al., 2018; ORUGANTI et al., 2016) Ao mesmo tempo, a capacidade dos métodos teóricos de fornecer uma compreensão profunda dos processos de emissão fotofísica e dos mecanismos de reação fotoquímica é melhor explorada ao permitir que outras propriedades dos estados ocupem o centro do palco, como suas geometrias de equilíbrio. No entanto, dados de referência confiáveis sobre, por exemplo, geometrias de estado excitado e frequências vibracionais necessárias para referências alternativas são escassos e não são facilmente obtidos por meios experimentais ou computacionais, exceto para moléculas pequenas. Embora as geometrias de estado excitado possam agora ser calculadas usando uma ampla variedade de métodos para os quais implementações eficientes de gradientes analíticos estão disponíveis (DELCEY et al., 2014), é de interesse especial avaliar o desempenho da Teoria do Funcional de Densidade Dependente do Tempo (TD-DFT) para tais cálculos, pois esta é a abordagem mais popular para a modelagem de estados excitados na química quântica atual (WANG; DURBEEJ, 2020). A investigação de um ponto estacionário das curvas de energia potencial da molécula CH3Cl evidenciou a formação de um par iônico estabilizado por uma ligação de hidrogênio intramolecular, H2C+ H•••Cl-. Esta estrutura foi mapeada pela primeira vez e corresponde a um novo tipo de ligação de hidrogênio (MEDEIROS et al., 2016). Outros trabalhos do grupo de pesquisa de Modelagem Computacional de Reações Atmosféricas, Orgânicas e de Interesse Biológico, estudaram a fotoquímica de outros haloalcanos, como CFCs e HCFCs, além da caracterização de suas geometrias de pares iônicos, os quais levam a formação de fragmentos carregados (como o Cl-) nos limites de dissociação (BEZERRA et al., 2021; MEDEIROS et al., 2018; RODRIGUES et al., 2016; RODRIGUES et al., 2019; SILVA, 2019; VENTURA; DO MONTE, 2020). A fim de obter uma compreensão teórica completa dos mecanismos fotoquímicos, é necessário determinar todas as importantes vias reacionais após a fotoexcitação. Desta forma, o objetivo deste trabalho é obter um funcional TD- DFT que reproduza melhor certas propriedades, como energias de excitação vertical e configurações eletrônicas de estados de valência e Rydberg, e a geometria otimizada do par iônico do CH2FCl, ambas no nível de referência, MR-CISD. Este método é considerado altamente útil e preciso na descrição de estados excitados, principalmente para sistemas pequenos (LISCHKA et al., 2018).
Material e métodos
Uma vez que este trabalho tem por objetivo obter um funcional o qual melhor descreve as características fotoquímicas no nível MR-CISD(+Q) (SILVA, 2019) utilizando uma geometria experimental (ver BLANCO et al., 1995), a estratégia adotada foi testar a performance de vários funcionais. Inicialmente, foram realizados cálculos single point TD-DFT na geometria experimental (BLANCO et al., 1995), com 19 estados singletos, com funções de bases d- aug-cc-pVDZ (C) e aug-cc-pVDZ (H, F, Cl). Os funcionais usados foram B3LYP, CAM-B3LYP, BHandHLYP, ωB97XD, PBE1PBE, M06 e M06-2X. Após obter as propriedades de interesse foi realizada uma caracterização dos estados eletrônicos excitados no tocante às configurações e energias eletrônicas, bem como uma comparação com os dados no nível de referência (MR-CISD+Q) na geometria experimental quanto às mudanças das propriedades. Nessa parte do trabalho foram analisadas as energias de excitação vertical, transições eletrônicas e os orbitais envolvidos nessas transições. Contudo, foi necessário aumentar o número de estados nos cálculos single point a fim de obter uma completa correspondência no assinalamento dos estados do método modelo. Aumentou-se essa quantidade para 31 (PBE1PBE), 32 (B3LYP), 35 (CAM-B3LYP), 40 (BhandHLYP e M06), 42 (ωB97XD) e 45 (M06-2X). Posteriormente, foi feita a caracterização dos estados TD-DFT novamente, seguida da comparação com os respectivos estados do metodo MR- CISD(+Q). Por último, fez-se a otimização e frequência (3 estados singletos e bases d-aug-cc-pVDZ (C)/aug-cc-pVDZ (H, F, Cl)) do par iônico formado no estado excitado do CH2FCl. Os cálculos TD-DFT foram realizados no programa Gaussian 09 (FRISCH et al., 2016).
Resultado e discussão
O benchmarking, investigação na qual os resultados de alguns métodos
menos precisos são comparados com um método mais robusto e de maior qualidade,
foi realizado comparando-se alguns funcionais TD-DFT com os resultados MR-
CISD+Q (SILVA, 2019). Os funcionais estudados foram B3LYP, CAM-B3LYP, ωB97XD,
BHandHLYP, PBE1PBE, M06 e M06-2X. Devido a sua eficiência para a descrição de
estados excitados (FERRÉ et al., 2016; LAURENT; JACQUEMIN, 2013;
LESZCZYNSKI, 2017; SILVA-JUNIOR et al., 2008), os funcionais aqui
citados foram escolhidos como candidatos para o benchmarking deste
trabalho. Um ponto importante a ser destacado nesta avaliação é que o método
TD-DFT, por ser single reference não irá reproduzir exatamente os
resultados MR-CISD+Q. O último fornece uma boa descrição tanto do estado
fundamental quanto dos excitados quanto às excitações de dois elétrons ou
interseções cônicas, enquanto o primeiro não possui essas qualidades (CARICATO
et al., 2010). Logo, não se espera que haja uma completa reprodução
simplesmente realizando um cálculo single point TD-DFT com as mesmas
coordenadas nucleares (geometria) e igual quantidade de estados (19) e funções
de base (d-aug-cc-pVDZ (C) e aug-cc-pVDZ (Cl, F e H)) que o MR-CISD(+Q). Mas é
necessário ter a melhor correspondência possível para que o funcional consiga
reproduzir corretamente o comportamento dos estados excitados, particularmente
os canais de formação do par iônico. Deste modo, a reprodução completa dos
estados TD-DFT paralela ao nível de referência se deu a partir da quantidade de
estados citada em Material e Métodos. Fez-se uma análise dos erros relativos
para cada estado TD-DFT análogo ao nível MR-CISD+Q. A Figura 1a) apresenta os
erros relativos (em %) entre os estados TD-DFT em relação aos resultados de
referência. Pode-se observar que para os estados n1-σCCl*
(11A' no método modelo) e n2-σCCl*
(21A" no método modelo) três funcionais apresentaram erros acima de
20%, sendo eles: BHandHLYP (26,4% e 27%, respectivamente), ωB97XD (23,4% e
23,2%, respectivamente) e CAM-B3LYP (ambos com 23,0%). Além disso, o BHandHLYP
atingiu o maior e o menor erros dentre toda a figura os quais foram,
respectivamente, 27% (estado n2-σCCl*) e 0,05%
(61A" no método modelo). O B3LYP apresentou erros mínimo de 7,6%
(21A' e 11A') e máximo de 20% (estado
n2-3pσ). Para o PBE1PBE esses erros foram de 5,5%
(71A") e 17,4% (41A"), respectivamente. O M06 apresentou,
em sua maioria, erros acima de 10%. Enquanto o M06-2X apresentou um erro máximo
de 6,0% (estados 11A" e 31A") e um erro mínimo de 2,8%
(estado nF--3pσ). Desta forma, conclui-se que o M06-2X
reproduziu melhor os dados do método de referência com relação às energias de
excitação vertical, pois foi o único funcional que apresentou baixos erros
relativos para todos os estados. Segundo Alipour (2019), o M06-2X descreve
muito bem estados excitados de valência. Enquanto Jacquemin et al.
(2010) mostrou que esse mesmo funcional relatou menores erros nas energias dos
estados de Rydberg comparado com PBE1PBE, B3LYP, CAM-B3LYP e M06. Na Figura 1b)
é mostrada a correlação entre as energias de excitação vertical obtidas com os
diferentes funcionais TD-DFT e MR-CISD+Q. Para simplificar o gráfico, as
energias MR-CISD+Q foram colocadas em ordem crescente, independente da simetria
dos estados correspondentes a elas, enquanto as energias dos funcionais
acompanharam a correspondência. Conforme pode ser observado, os maiores desvios
foram obtidos para os funcionais M06 e B3LYP. Em geral, o BHandHLYP foi o
funcional com os melhores resultados, com os maiores desvios para os dois
primeiros e dois últimos estados excitados. Todos os estados se encontram na
faixa abaixo de 12,5 eV. Além disso, a natureza dos estados foi relativamente
bem descrita por todos os funcionais usados para os cálculos TD-DFT, mas com um
número muito maior de estados. Uma análise também importante diz respeito a
descrição da estrutura de mínimo observada no estado eletrônico S3.
Essa estrutura já foi observada em diversos haloalcanos, entre eles CFCs e
HCFCs, estudados pelo nosso grupo e também existe no caso do 1-cloro-1-flúor-
metano (SILVA, 2019). Trata-se de uma estrutura iônica na qual há a presença de
uma ligação de hidrogênio intramolecular, HFC+H•••Cl-.
Esta foi obtida através de cálculos de otimização completa de geometria
utilizando o método MR-CISD e a base aug-cc-pVTZ para o sistema sem simetria
com 3 estados eletrônicos (21A' + 11A'') e espaço ativo
formado apenas com os orbitais de valência envolvidos na ligação C−Cl (CAS
(6,4): n1 (Cl), n2 (Cl), nσ (Cl) e
2pσ (C). O que se pretende aqui é investigar se os funcionais TD-DFT
reproduzem satisfatoriamente essa estrutura. Para todos os métodos foram
realizados cálculos de otimização completa de geometria, seguido do cálculo de
frequência. A Figura 2 mostra as geometrias para o estado iônico tanto a nível
TD-DFT quanto no método de referência. Apenas o funcional CAM-B3LYP não gerou
uma geometria de mínimo, mas um estado de transição (TS). Para todas as
arquiteturas moleculares da Figura 2 todos os átomos constituintes (C, Cl, F e
H) de uma mesma estrutura estão totalmente ou quase coplanares. A fim de
facilitar a explicação, o H mais distante do Cl em cada estrutura será chamado
H’. Nota-se que para o B3LYP o cloro se encontra entre os átomos de F e H, onde
RH-Cl = 2,43 Å, 0,43 Å maior do que a respectiva ligação no nível
MR-CISD(+Q). Por outro lado, ambas divergem apenas 0,01 Å na distância de
ligação C−F. As maiores diferenças entre esses métodos ocorrem para o
<CHCl e RC-H’, os quais são 30,1° e 0,69 Å,
respectivamente. O PBE1PBE mostrou uma singularidade em relação às outras
arquiteturas TD-DFT. O Cl apareceu entre os dois átomos de H, a qual RH-
Cl = 1,81 Å com o átomo de hidrogênio mais próximo, cerca de 0,2 Å menor
que a mesma distância no método modelo. Neste, as diferenças correspondentes
com o PBE1PBE são <CHCl = 28,9°, RF-C = 0,03 Å e no
mínimo 0,13 Å para as distâncias C−H. Logo, devido a essas diferenças com a
geometria do estado iônico MR-CISD(+Q) os dois funcionais citados há pouco
foram considerados de qualidade ruim. Por conta da ausência de correspondência
dos estados 81A' e 101A' (respectivamente,
σCCl-3pσ e σCCl-σCCl*) e da grande
diferença da distância de ligação H−Cl (RH-Cl = 2,20 Å) em relação
ao nível MR-CISD(+Q) (RH-Cl = 2,0 Å), o BHandHLYP foi também
descartado para boa performance dos dados no nível modelo. No caso do M06 e do
M06-2X há boa concordância com os dados geométricos obtidos com o método de
referência. O M06 apresenta a mesma distância de ligação H−Cl (2,0 Å), enquanto
no M06-2X essa distância é 0,12 Å maior que a geometria a nível MR-CISD(+Q).
Por outro lado, as diferenças nas distâncias de ligação C−H e C−H’ são menores
a nível M06-2X do que o funcional M06 quando comparadas com a da referência,
sendo, respectivamente, 0,03 Å e 0,04 Å para o primeiro e 0,06 Å e 0,06 Å para
o segundo funcional. Outro parâmetro importante é o ângulo CHCl, o qual
observa-se a maior semelhança para o M06 (179,6°) com a geometria de referência
(171,5°), apesar deste parâmetro geométrico, a estrutura M06-2X mostrou
relativa similaridade com a referência e melhor desempenho com relação às
energias de excitação vertical e na natureza dos estados com relação ao MR-
CISD+Q.
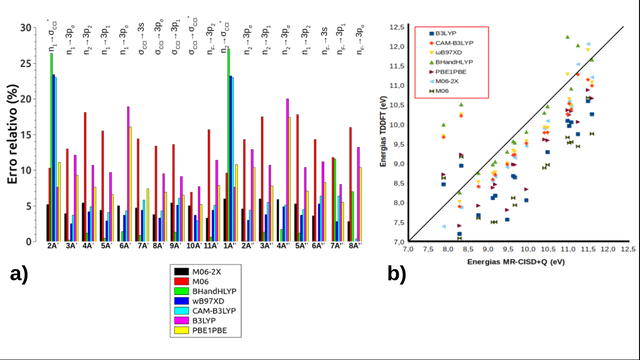
Erros relativos (a) e Correlação (b) das energias dos estados TD-DFT com as respectivas energias dos estados MR-CISD+Q.
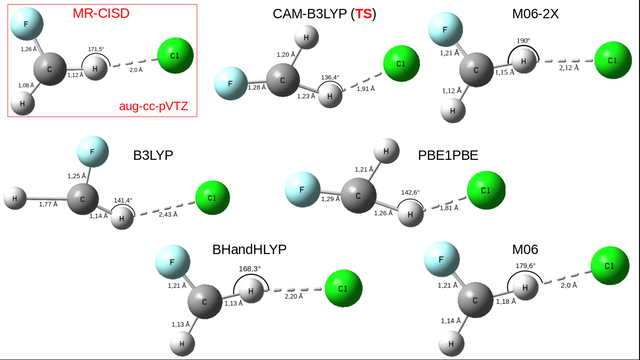
Geometrias otimizadas do par iônico do HCFC-31 com os métodos TD-DFT/d-aug-cc-pVDZ (C)/aug-cc-pVDZ (F, Cl, H) e MR-CISD/aug-cc-pVTZ (ver SILVA, 2019).
Conclusões
É importante validar aproximações práticas de funcionais de densidade para verificar a confiabilidade de suas energias de excitação e transições eletrônicas previstas. Neste trabalho foi feito um estudo de aferição o qual avaliou os funcionais B3LYP, CAM-B3LYP, ωB97XD, BHandHLYP, PBE1PBE, M06 e M06- 2X a nível TD-DFT usando as bases d-aug-cc-pVDZ (C) e aug-cc-pVDZ (Cl, F e H). Nenhum deles, seja da família M06 ou qualquer outro funcional considerado, mostrou exatidão aceitável para os dois tipos de excitações de valência e Rydberg utilizando 19 estados. Contudo, após o aumento desse número nos cálculos single-point, houve uma boa correspondência dos estados TD-DFT com os respectivos estados MR-CISD(+Q). Os erros relativos na análise das energias de excitação vertical apontaram que o TD-M06-2X apresentou erros abaixo de 6% para todos os estados, sendo considerado neste quesito aquele funcional que melhor reproduziu as energias do método modelo. Enquanto funcionais como o ωB97XD, BHandHLYP e CAM-B3LYP apresentaram valores acima de 20%. Embora tenha ocorrido uma boa correlação da maioria das energias de excitação eletrônica do TD-BHandHLYP, este não apresentou os estados com configuração σCCl-3pσ (81A') e σCCl- σCCl* (101A'). As geometrias de mínimo referentes ao estado de par iônico obtidas com TD-DFT e funções de bases d-aug-cc-pVDZ (C)/aug-cc-pVDZ (Cl, F e H) demonstraram que os funcionais BHandHLYP, M06 e M06-2X apresentaram maiores semelhanças com a respectiva estrutura a nível MR- CISD/aug-cc-pVTZ. Apesar do alto valor do ângulo CHCl (190°), a estrutura do par iônico M06-2X mostrou relativa similaridade com os outros parâmetros geométricos da geometria de referência e o melhor desempenho com relação às energias de excitação vertical e na natureza dos estados com relação ao MR- CISD+Q.
Agradecimentos
Todos os autores agradecem às agências de fomento CAPES, CPNq e FAPESQ pelos apoio financeiro, à UFPB, e especialmente, à todos do Laboratório de Modelagem Molecular de Reações Químicas (LMMRQ).
Referências
ABAS, N.; KALAIR, A. R.; KHAN, N.; HAIDER, A.; SALEEM, Z.; SALEEM, M. S. Natural and synthetic refrigerants, global warming: A review. Renewable and Sustainable Energy Reviews, v. 90, p. 557-569, 2018.
ALIPOUR, M. Theoretical prediction of valence and Rydberg excited states: Minnesota exchange- correlation functionals vs symmetry adapted cluster-configuration interaction. International Journal of Quantum Chemistry, e25898, 2019.
BHATNAGAR, A.; CARR, R. W. HCFC-31: the CHClFO2 + NO → CHClFO + NO2 reaction and Cl atom elimination from CHClFO. Chemical Physics Letters, v. 258, p. 651-656, 1996.
BEZERRA, M. G.; LEITÃO, E. F. V.; ANDRADE, R. B.; VENTURA, E.; ANDRADE, DO MONTE, S. A. Photochemistry of Monohydrated Chloromethane: Formation of Free and Hydrated Cl- and CH3+ Ions from a Solvent-Shared Semi-Ion-Pair. The Journal of Physical Chemetry A, v. 125, p. 8603−8614, 2021.
BLANCO, S.; LESARRI, A.; LOPEZ, J. C.; ALONSO, J. L.; GUARNIERI, A. The Rotational Spectrum of Chlorofluoromethane. Journal of Molecular Spectroscopy, v. 174, p. 397–416, 1995.
CARICATO, M.; TRUCKS, G.W.; FRISCH, M. J.; WIBERG, K. B. Electronic Transition Energies: A Study of the Performance of a Large Range of Single Reference Density Functional and Wave Function Methods on Valence and Rydberg States Compared to Experiment. Journal of Chemical Theory and Computation, v. 6, p. 370–383, 2010.
CHARMET, A. P.; STOPPA, P.; TASINATO, N.; GIORGIANNI, S.; BARONE, V.; BICZYSKO, M.; BLOINO, J.; CAPPELLI, C.; CARNIMEO, I.; PUZZARINI, C. An integrated experimental and quantum- chemical investigation on the vibrational spectra of chlorofluoromethane. The Journal of Chemical Physics, v. 139, n. 164302, 2013.
DELCEY, M.G. ; FREITAG, L.; PEDERSEN, T. B.; AQUILANTE, F.; LINDH, R.; GONZÁLEZ, L. Analytical gradients of complete active space self-consistent field energies using Cholesky decomposition: Geometry optimization and spin-state energetics of a ruthenium nitrosyl complex. The Journal of Chemical Physics, v. 140, 2014.
ESSA, A. G. A.; MOHAMED, A. A. Selection of optimal fluid for refrigeration cycles. World Journal of Advanced Engineering Technology and Sciences, v. 1, p. 21–36, 2020.
FERRÉ, N.; FILATOV, M.; HUIX-ROTLANT, M. Density-Functional Methods for Excited States, Springer: [s.l.], 2016.
FRISCH, M. J.; TRUCKS, G. W. ; SCHLEGEL, H. B.; SCUSERIA, G. E.; ROBB, M. A.; CHEESEMAN, J. R.;SCALMANI, G.; BARONE, V.; PETERSSON, G. A.; NAKATSUJI, H.; LI, X.; CARICATO, M.; MARENICH, A.; BLOINO, J.; JANESKO, B. G.; GOMPERTS, R.; MENNUCCI, B.; HRATCHIAN, H. P.; ORTIZ, J. V.; IZMAYLOV, A. F.; SONNENBERG, J. L.; WILLIAMS-YOUNG, D.; DING, F.; LIPPARINI, F.; EGIDI, F.; GOINGS, J.; PENG, B.; PETRONE, A.; HENDERSON, T.; RANASINGHE, D.; ZAKRZEWSKI, V. G.; GAO, J.; REGA, N.; ZHENG, G.; LIANG, W.; HADA, M.; EHARA, M.; TOYOTA, K.; FUKUDA, R.; HASEGAWA, J.; ISHIDA, M.; NAKAJIMA, T.; HONDA, Y.; KITAO, O.; NAKAI, H.; VREVEN, T.; THROSSELL, K.; MONTGOMERY JR., J. A.; PERALTA, J. E.; OGLIARO, F.; BEARPARK, M.; HEYD, J. J.; BROTHERS, E.; KUDIN, K. N.; STAROVEROV, V. N.; KEITH, T.; KOBAYASHI, R.; NORMAND, J.; RAGHAVACHARI, K.; RENDELL, A.; BURANT, J. C.; IYENGAR, S. S.; TOMASI, J.; COSSI, M.; MILLAM, J. M.; KLENE, M.; ADAMO, C.; CAMMI, R.; OCHTERSKI, J. W.; MARTIN, R. L.; MOROKUMA, K.; FARKAS, O.; FORESMAN, J. B.; FOX, D. J., Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2016.
JACQUEMIN, D.; PERPÈTE, E. A.; CIOFINI, I.; ADAMO, C.; VALERO, R.; ZHAO, Y.; TRUHLAR, D. G. On the Performances of the M06 Family of Density Functionals for Electronic Excitation Energies. Journal of Chemical Theory and Computation, v. 6, p. 2071–2085, 2010.
LAURENT, A. D.; JACQUEMIN, D. TD-DFT Benchmarks: A Review. International Journal of Quantum Chemistry, v. 113, p. 2019–2039, 2013.
LESZCZYNSKI, J. Handbook of Computational Chemistry. Springer: [s.l.], 2 ed., 2017.
LISCHKA, H.; NACHTIGALLOVÁ, D.; AQUINO, A. J. A.; SZALAY, P. G.; PLASSER, F.; MACHADO, F. B. C.; BARBATTI, M. Multireference Approaches for Excited States of Molecules. Chemical Reviews, v. 118, p. 7293-7361, 2018.
LOOS, P. F.; SCEMAMA, A.; BLONDEL, A.; GARNIRON, Y.; CAFFAREL, M.; JACQUEMIN, D. A Mountaineering Strategy to Excited States: Highly Accurate Reference Energies and Benchmarks. Journal of Chemical Theory and Computation, v. 14, p. 4360-4379, 2018.
MEDEIROS, V. C.; ANDRADE, R. B.; LEITÃO, E. F. V.; VENTURA, E.; BAUERFELDT, G. F.; BARBATTI, M.; DO MONTE, S. A. Photochemistry of CH3Cl: Dissociation and CH•••Cl Hydrogen Bond Formation. Journal of the American Chemical Society, v. 138, p. 272-280, 2016.
MEDEIROS, V. C.; ANDRADE, R. B.; RODRIGUES, G. P.; VENTURA, E.; BAUERFELDT, G. F.; BARBATTI, M.; DO MONTE, S. A. Photochemistry of CF3Cl: Quenching of Charged Fragments Is Caused by Nonadiabatic Effects. Journal of Chemical Theory and Computation, v. 14, p. 4844−4855, 2018.
MOLINA, M. J.; ROWLAND, F. S. Stratospheric sink for chlorofluoromethanes: chlorine atomic-atalysed destruction of ozone. Nature, v. 249, p. 810-812, 1974.
ORUGANTI, B.; FANG C.; DURBEEJ, B. Assessment of a composite CC2/DFT procedure for calculating 0–0 excitation energies of organic molecules. Molecular Physics, v. 114, p. 3448-3463, 2016.
RODRIGUES, G. P.; VENTURA, E.; DO MONTE, S. A.; BARBATTI, M. UV-Photoexcitation and Ultrafast Dynamics of HCFC-132b (CF2ClCH2Cl). Journal of Computational Chemistry, v. 37, p. 675-683, 2016.
RODRIGUES, G. P.; LIMA, T. M. L.; ANDRADE, R. B.; VENTURA, E.; DO MONTE, S. A.; BARBATTI, M. Photoinduced Formation H-Bonded Ion Par in HCFC-133a. The Journal of Physical Chemistry A, v. 123, p. 1953-1961, 2019.
SILVA, Albert Jorgivan Filho Wesley Hess de Sousa Silva. Estudo Teórico da Fotoquímica do 1- cloro-1-flúor-metano (HCFC-31. 2019. 77 f. Dissertação (Mestrado em Química) – Departamento de Química, Universidade Federal da Paraíba, João Pessoa, 2019.
SILVA-JUNIOR, M. R.; SCHREIBER, M.; SAUER, S. P. A.; THIEL, W. Benchmarks for electronically excited states: Time-dependent density functional theory and density functional theory based multireference configuration interaction. The Journal of Chemical Physics, v. 129, p. 104103, 2008.
SHANKLIN, J. Reflections on the ozone hole. Nature, v. 465, p. 34-35, 2010.
VENTURA, E.; DO MONTE, S. A. Hydrogen‐bonded contact ion pair in gaseous chloroethane: a multi‐reference configuration interaction with singles and doubles (MR‐CISD) study including extensivity corrections, Theoretical Chemistry Accounts, v. 139, 2020.
WANG, J.; DURBEEJ, B. How accurate are TD-DFT excited-state geometries compared to DFT ground-state geometries? Journal of Computational Chemistry, p. 1-12, 2020.