ÁREA
Iniciação Científica
Autores
Santos, P.L.L. (UEMA) ; Almeida, P.H.B. (UEMA) ; Viana, K.P. (UEMA) ; Sousa, A.S. (UEMA) ; Fernandes, R.M.T. (UEMA) ; Khan, A. (UEMA)
RESUMO
Neste estudo, investigamos as características termodinâmicas para determinar os parâmetros de taxa de reação de dissociação e abstração durante a decomposição da molécula Dinitrodimetiloxamida. A partir dos resultados obtidos, analisamos as informações termodinâmicas para avaliar os constantes de velocidades nas formações de radicais da molécula de interesse. Observamos que, os constantes de velocidades para a decomposição térmica primário (ddodpt1) e secundário (ddodpt2) aumenta com temperatura e no caso de radicais terciário (ddodt3) e quaternário (ddodt4) demostra comportamento inversa em relação a temperatura. Assim, podemos concluir que a iniciação de reação de combustão Dinitrodimetiloxamida depende nas abstrações de hidrogênio primário e secundário.
Palavras Chaves
Termodinâmica; Decomposição térmica ; DFT
Introdução
O conhecimento dos aspectos energéticos das moléculas desempenha um papel fundamental em diversas áreas da ciência e tecnologia, pois contribui significativamente para o estudo das relações complexas entre energia, estrutura e reatividade. Ao interpretar os parâmetros termoquímicos, especialmente os valores de energia livre de Gibbs, podemos obter uma compreensão mais aprofundada da natureza das ligações químicas. Isso nos permite obter informações valiosas sobre a energia envolvida nessas ligações, além de fornecer suporte para correlacionar características estruturais e reacionais dos compostos que contêm essas ligações (SILVA, 2001). Uma vez que a estrutura tenha sido elucidada, o aspecto mais crucial a ser computado é a energia inerente à molécula. A menos que se trate de moléculas completamente inflexíveis, a conformação da estrutura se dispersa por uma série de arranjos configuracionais nos quais a está intimamente ligada à energia envolvida. Portanto, para a avaliação de qualquer propriedade, inicialmente deve estabelecer tanto as energias relativas das diversas conformações quanto a identificação daquela que apresenta maior estabilidade. A quantificação energética reflete a disparidade energética da molécula (ou átomo) em relação a um estado estacionário definido por elétrons e núcleo de forma isolada, o qual é considerado como referencial de energia zero. Assim, os valores energéticos atribuídos às espécies sob análise assumem valores negativos e indicam a energia necessária para a completa dissociação dos elétrons e núcleo, ou alternativamente, a energia liberada quando os elétrons e núcleos se amalgamam para dar origem à espécie (ORTALAN, 2014). A energia livre de Gibbs (G) é uma medida conveniente para avaliar o desvio de uma reação de seu ponto de equilíbrio quando reagentes e produtos estão em seu estado de referência. Dessa forma, podemos inferir que uma reação química será sempre espontânea quando ela for exotérmica (∆H < 0) e resultar em aumento de entropia (∆S > 0), pois isso leva a um valor negativo de ∆G, indicando que a reação é termodinamicamente espontânea. Ao reunir os resultados de todas as moléculas, calculamos as variações da energia livre de Gibbs (∆G) utilizando a diferença entre os produtos e reagentes. Essa abordagem nos permite compreender melhor o comportamento termodinâmico das reações em estudo e determinar sua espontaneidade (LIMA, 2018). A fórmula molecular C4H6N4O6, conhecida como Dinitro Dimethyl Oxamide, possui dois grupos metil em sua estrutura, que podem ser considerados fontes de hidrogênio, enquanto há uma quantidade suficiente de oxigênio ativo. Através da análise termoquímica e cinética da decomposição térmica dessas matérias de alta energia, buscamos obter informações sobre suas propriedades, ligações energéticas e comportamentos em diferentes temperaturas (ALIEV, 2016). O cálculo da constante de velocidade é muito mais complicado que os cálculos da constante de equilíbrio ou quantidades como a entalpia, energia livre ou entropia, por exemplo. Tanto a constante de velocidade como a constante de equilíbrio estão relacionado com a diferença energética entre duas espécies: a constante de velocidade entre os reagentes e o estado de transição, e a constante de equilíbrio entre os produtos e reagentes. Assim, a constante de velocidade, K, pode ser aproximada, para reações de primeira ordem (LEWARS, 2011). O objetivo desse trabalho foi realizar uma avaliação decomposição térmica das matérias de alta energia para determinar as propriedades termodinâmicas das várias estruturas intermediarias da mesma, buscando a descrever um mecanismo da reação elementar para a reação de combustão. Para alcançar os nossos objetivos, utilizamos o método teórico de DFT com função de base cc-pVDZ para realizar os cálculos, nas temperaturas com intervalos de 5, 298, 500 e 1000K.
Material e métodos
As geometrias das estruturas de radicais e moléculas criadas pelo programa GaussView (TOMBERRG, 2013). Levando em consideração os comprimentos de ligação entre N-N, NC, N-O, C-C, C-O e C-H, foram otimizadas utilizando o método de DFT (B3LYP) com função de base cc-pVDZ, nas temperaturas de 5K, 298K, 500K e 1000K. Após otimização, a matriz hessiana e modos normais das mesmas foram calculadas utilizando o método B3LYP/cc-pVDZ nas temperaturas de mencionadas (BURKE; WAGNER, 2013). Após realizar todos os cálculos, os resultados da energia livre de Gibbs foram cuidadosamente analisados, e a partir deles, procedeu-se ao cálculo da constante de velocidade (K). Essa constante desempenha um papel crucial na determinação da taxa de decomposição e na velocidade das reações envolvidas, o que impacta diretamente no comportamento do sistema em estudo (OCHTERSKI, 2000). No presente estudo, a molécula Dinitro Dimethyl Oxamide (ddo) passa por uma decomposição térmica (encontra-se abaixo em figura - 1), resultando nas estruturas ddodpt1, ddodpt2, ddodpt3 e ddodpt4, por meio da remoção de átomos específicos da estrutura original. Inicialmente, um átomo de hidrogênio é removido da molécula principal, resultando na formação da molécula ddodpt1. Em seguida, a remoção de CH2 resulta na molécula ddodpt2. Da molécula ddodpt2, é removido o NO2, formando a molécula dddopt3. A molécula ddodpt3 é quebrada na ligação entre os átomos de carbono, resultando na formação da molécula ddodpt4. Cada molécula presente na decomposição térmica levou em torno de quatro horas e quarenta minutos para calcular sua otimização e frequência. Em especial as moléculas Dinitro Dimethyl Oxamide que levou um custo computacional até cinco horas e cinquenta minutos. Todos os cálculos foram realizados utilizando o computador com configuração de um processador Intel Core i5 de quinta geração, uma placa gráfica GTX 1080, 8 GB de RAM, um SSD de 230 GB e uma placa-mãe ASUS DDR4.
Resultado e discussão
A decomposição térmica é um processo químico no qual uma molécula se decompõe em
outros radicais quando submetida a altas temperaturas. Durante essa reação, a
energia térmica é fornecida para quebrar as ligações químicas dentro da molécula,
resultando na formação de produtos diferentes dos reagentes originais. Observa-se os
resultados, A energia livre de Gibbs revela um aumento progressivo à medida que a
temperatura se eleva, como evidenciado nos valores da molécula ddo é -26,982,
-57,103, 156,887 kJ/mol, respectivamente. Importante notar que todos os resultados
calculados exibem ΔG inferior a zero para todas as temperaturas analisadas. As
constantes de velocidade ddodpt1 e ddodpt2 revelam um aumento notável na taxa de
decomposição à medida que a temperatura ascende. Mais especificamente, a constante
ddodpt1 registra um incremento de 2,93 vezes ao evoluir de 298K para 500K, seguido
por um salto de 4,14 vezes entre 500K e 1000K. De maneira similar, a constante
ddodpt2 apresenta um acréscimo de 1,91 vezes entre 298K e 500K, e um aumento ainda
mais pronunciado de 5,68 vezes de 500K a 1000K.
Com esse padrão, as constantes ddodpt3 e ddodpt4 exibem valores consideravelmente
superiores a 298K, entretanto, diminuem à medida que a temperatura se eleva. Essa
tendência peculiar é atribuída à estabilidade intrínseca da molécula, que se
manifesta de maneira proeminente em temperaturas mais baixas. Consequentemente, a
decomposição ocorre de forma mais célere em intervalos térmicos mais baixos. Assim,
as constantes ddodpt3 e ddodpt4 sugerem uma decomposição mais rápida a 298K,
contrastando com ddodpt1 e ddodpt2, que indicam uma decomposição mais eficaz a
1000K. Segundo o Chase (1998), os valores de entalpia de NO2 exibem variação entre
298K e 1200K, totalizando 33,10 kJ/mol. Contudo, por meio de cálculos computacionais
(DFT, B3LYP) no presente estudo, obtivemos um resultado de 42,67 kJ/mol na mesma
faixa de temperatura. Essa discrepância pode ser imputada ao fato de que a molécula
em análise se encontra isolada no vácuo durante o cálculo por meio do programa
Gaussian. Além disso, a natureza computacional do experimento pode gerar desvios em
relação aos resultados empíricos.
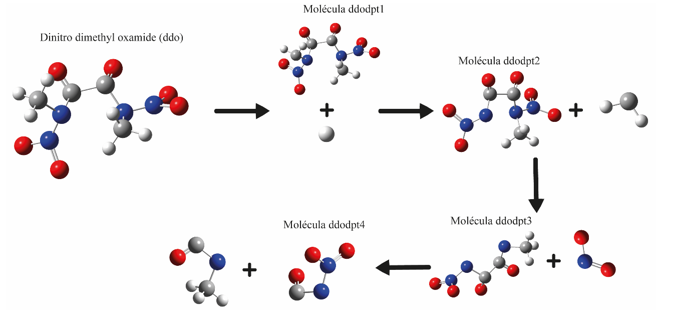
Decomposição térmica da molécula de \r\nDinitrodimetiloxamida.
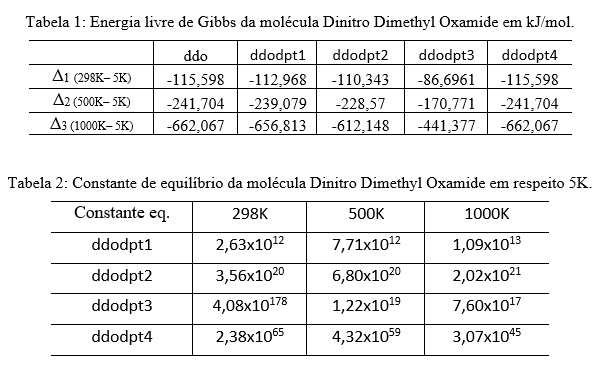
Constante de equilíbrio da molécula Dinitro Dimethyl \r\nOxamide em respeito 5K e Energia livre de Gibbs da \r\nmolécula Dinitro Dimethyl Oxamide em kJ/mol.
Conclusões
Para obter resultados otimizados na decomposição da Dinitro Dimethyl Oxamide e acelerar a formação de produtos, é recomendável um processo que inicie em temperaturas elevadas e, progressivamente, reduza a temperatura à medida que a decomposição avança. Esse procedimento capitaliza o aumento de velocidade das constantes ddodpt1 e ddodpt2 em temperaturas mais altas, enquanto explora a estabilidade e rapidez de decomposição das constantes ddodpt3 e ddodpt4 em temperaturas mais baixas. Tais conclusões ressaltam a relevância vital de compreender e aplicar as constantes de velocidade para aprimorar processos químicos, garantindo, assim, uma decomposição mais eficiente e produtiva.
Agradecimentos
À UEMA e FAPEMA pela concessão da bolsa e pelo fomento da pesquisa. Ao Grupo de estudos do Laboratório de Físico-Química da UEMA. Ao programa de Iniciação Científica – PIBIC/UEMA e ao professor Alamgir Khan.
Referências
ALIEV, Z. G. et al. Estrutura e propriedades de um cristal bimolecular (2CL-20+ MNO). Journal of Structural Chemistry, v. 57, n. 8, p. 1613-1618, 2016.
CHASE, M. W.; NATIONAL INFORMATION STANDARDS ORGANIZATION (US). NIST-JANAF thermochemical tables. Washington, DC: American Chemical Society, 1998.
LEWARS, E. G. Computational chemistry. Introduction to the theory and applications of molecular and quantum mechanics, v. 318, 2011.
LIMA, N. E. M. Comparação de métodos computacionais para estimar a energia livre de Gibbs em reações bioquímicas. Tese de Doutorado - Universidade do Minho Portugal, 2018.
MAHAN, B. M.; MYERS, R. J.; Química um curso universitário, 4ª edição, Edgard Blücher, 1996.
ORTOLAN, Alexandre Osmar. Apostila de práticas de química computacional. Trabalho de Conclusão de Curso. Universidade Tecnológica Federal do Paraná. 2014.
SILVA, M. D. C. R. Energética de Complexos Metálicos, 2001.
TOMBERG, A. Gaussian 09w tutorial. An introduction to computational chemistry using G09W and Avogadro software, p. 1-36, 2013.
TOMBERG, A. Gaussian 09w tutorial. An introduction to computational chemistry using G09W and Avogadro software, p. 1-36, 2013.
BURKE, K.; WAGNER, L. O. DFT in a nutshell. International Journal of Quantum Chemistry, v. 113, n. 2, p. 96-101, 2013.
OCHTERSKI, J. W. Thermochemistry in gaussian. Gaussian Inc, v. 1, p. 1-19, 2000.