ÁREA
Química Medicinal
Autores
Viegas da Silva, A. (UNIFAP) ; Luis Belém dos Santos, K. (UNIFAP) ; de Fátima Mendes Monteiro, B. (UNIFAP) ; dos Santos Borges, R. (UFPA) ; Breno Rodrigues dos Santos, C. (UNIFAP)
RESUMO
A Química Medicinal e a tecnologia computacional otimizam o desenvolvimento de fármacos, uma vez que possibilita a obtenção in sílico das propriedades químicas, eletrônicas, físico-químicas e farmacológicas de moléculas. Este estudo usa cálculos quânticos no método Density Functional Theory (DFT) na base B3LYP/6-31g(d,p) para analisar um novo antibiótico, com bioatividade bactericida. Descritores químicos quânticos são excelentes norteadores para a busca de novos candidatos a antimicrobianos para enfrentar a problemática da resistência bacteriana. A Química Computacional é uma poderosa ferramenta que possibilita triar novos compostos em bases de dados virtuais, impulsionando o desenvolvimento de novos fármacos e sendo eficiente com recursos para Pesquisa, Desenvolvimento e Inovação.
Palavras Chaves
Bioinformática; Química Medicinal; Fármacos
Introdução
A Química Medicinal desempenha um papel fundamental na identificação de fármacos, combinando a compreensão das bases moleculares da ação dos fármacos com ferramentas da química, farmácia, biotecnologia e tecnologia computacional. Essa abordagem possibilita estudos precisos de interações biológicas, otimizando tempo, recurso financeiro e experimentos biológicos dispendiosos com insumos tóxicos ou compostos instáveis, uma vez que as técnicas computacionais possibilitam a elucidação de propriedades físico-químicas potencialmente tóxicas ou carcinogênicas (COHEN, 1990; IMMING, 2008; SANTOS et al, 2014). O avanço do poder computacional impulsionou a modelagem molecular e estudos dos sistemas biológicos (RODRIGUES, 2001). Métodos computacionais permitem compreender, simular, explicar e prever o comportamento de moléculas bioativas, obtendo dados sobre suas propriedades geométricas, moleculares e estruturais, descritores fundamentais para entender a reatividade e estabilidade química de um composto (BORGES, 2017). A química computacional permite estimar as estabilidades moleculares de forma teórica, possibilitando entender e predizer o comportamento de uma molécula em sistemas biológicos, avaliando seu efeito farmacológico (BARREIRO et al, 1997; SILVERSTEIN; WEBSTER; KIEMLE, 2005; SOUZA; FARIAS, 2007;). Por conseguinte, o objetivo desde trabalho foi a aplicação de cálculos químicos- quânticos sobre a estrutura do composto QNZ, um novo antibiótico de efeito bactericida em cepas de Staphylococcus aureus resistentes a meticilina (MRSA), identificado por Bouley et al, (2015) assim, através da elucidação das propriedades de interesse químico e farmacológico, novas entidades químicas com potencial efeito bactericida possa ser identificado com uma abordagem da Química Computacional.
Material e métodos
A estrutura 2D com composto QNZ foi retirada do trabalho de Bouley, et al, (2015). Posteriormente a estrutura foi desenhada e pré otimizada em 3D no software ChemSketch 2012 (ACD LABS, 2012). No Software HyperChem 8.0.8 foi feita a otimização com o método de Molecular Mechanics na base MM+. Por fim, foi calculado as superfícies de energia potencial, ao qual combina-se os giros dos diedros com o cálculo de energia da geometria, procedimento este chamado de scan. Esta etapa final foi realizada no software Gaussian 09W, com o Método DFT, base B3LYP/6-31g(d,p) (FRISCH et al, 2009; NEGRELLI, 2011). Os diedros selecionados estão especificados na Figura 1. Foram realizadas 12 rotações de 30º nos diedros. O ambiente de cálculo foi o Sistema Operacional Windows 11, AMD Ryzen 9 3900X 12N/24T - 4.60Ghz, Memória Ram 32Gb 3200Mhz. O tempo de cálculo foi de aproximadamente 2 dias e 12 horas. Os resultados obtidos via Gaussian 09W (FRISCH et al, 2009) foram visualizados na interface gráfica GaussView 6.0, possibilitando obtenção dos dados acerca da Energia de Formação das conformações e menor e maior energia. O cálculo do root-meansquare deviation (RMSD) foi feito no software Discovery Studio Visualizer (BIOVITA FOUNDATION, 2007), realizando uma sobreposição da conformação bioativa cristalográfica com as conformações de menor e maior energia do composto QNZ após o cálculo de Scan.
Resultado e discussão
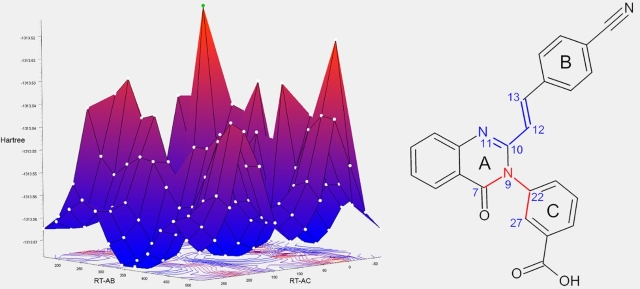
A Figura 1 ilustra as conformações espaciais calculadas para o composto em
estudo e as representações dos diedros que foram rotacionados.
A conformação geométrica de maior e menor energia possuem Energia de Formação de
-13,1351x10-2 e -13,1356x10-2 Hartree, respectivamente. Cada conformação
geométrica no Gráfico apresentará uma energia de formação variando entre os dois
extremos. Essa variação nas diferentes conformações deve-se aos diferentes
fatores eletrostáticos e estéricos, uma vez que interferem diretamente nos
equilíbrios das forças entre os átomos (SANTOS, 2021). A força intramolecular
não é uniforme, o compartilhamento de elétrons em uma molécula cria polos com
intensidade variando de acordo com a eletronegatividade dos átomos, sendo assim,
uma região molecular muito negativa tende a repelir outra região negativa, como
consequência, a conformação geométrica e a energia de formação irão variar
(ZUMDAHL; ZUMDAHL, 2007).
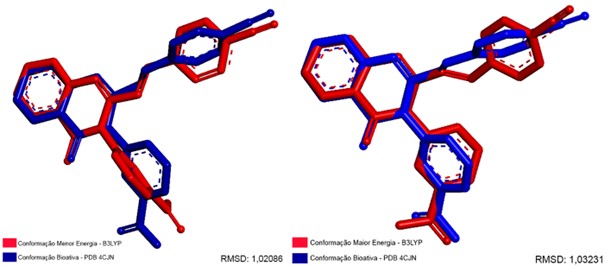
As conformações de Menor e Maior Energia foram comparadas com a conformação
Bioativa, observa-se um RMSD de 1,02 Å e 1,03 Å, respectivamente, conforme
Figura 2.
O RMSD mede o desvio entre as conformações. O valor é considerado aceitável
quando o RMSD < 2 Å (BORGES et al, 2019). Essa variação do RMSD nas conformações
é esperada e está fundamentada na hipótese que ligantes devem possuir regiões
flexíveis, possibilitando rotações e conformações moleculares para as interações
com os resíduos de aminoácidos dos sítios ativos de alvos biológicos, essa
capacidade flexível é o fator central que viabiliza a interação intermolecular
sob o sítio ao qual o ligante interage, um ligante muito “rígido” não
apresentará boa afinidade molecular nem as interações viáveis com os aminoácidos
que desencadearia o efeito farmacológico esperado (VERLI; BARREIRO, 2005).
Conclusões
Os descritores químicos quânticos norteiam busca por novas entidades bioativas, similares a compostos bioativos conhecidos. Virtual Screening em bases de dados na Bioinformática permite triagem de compostos com potencial atividade antimicrobiana. Os Cálculos da molécula estudada mostram flexibilidade relativa e baixa variação de energia, fornecendo dados para triagem de novos candidatos a fármacos eficazes contra bactérias multirresistentes em banco de dados de compostos conhecidos. Química Computacional é poderosa ferramenta no enfrentamento global deste problema.
Agradecimentos
Agradecer ao LMQC/UNIFAP pela orientação e ao NESBIO/UFPA pela Licença do Software Gaussian que permitiu rodar os cálculos químico-quânticos.
Referências
ACD Labs. Chemsketch Freware, versão 12.0. Toronto, 2012.
BARREIRO, E.; RODRIGUES, C. R.; ALBUQUERQUE, M. G.; Sant'Anna, C. M. R.; ALENCASTRO, R. B. Modelagem Molecular: Uma Ferramenta para o Planejamento Racional de Fármacos em Química Medicinal. Química Nova, v.20, n.3, p. 1-11, 1997. https://doi.org/10.1590/S0100-40421997000300011
BIOVITA FOUNDATION. Discovery Studio 4.0. San Diego: Accelrys, 2007.
BORGES, R. S.; PALHETA, I. C.; OTA, S. S. B.; MORAIS, R. B.; BARROS, V. A.; RAMOS, R. S.; SILVA, R. C.; COSTA, J. S.; SILVA, C. H. T. P.; CAMPOS, J. M.; SANTOS, C. B. R. Toward of Safer Phenylbutazone Derivatives by Exploration of Toxicity Mechanism. Molecules, 24, 143, 2019. DOI: 10.3390/molecules24010143.
BORGES, R. S.; Química Farmacêutica Fundamental. Belém: Universidade Federal do Pará, 2017.
BOULEY, R. et al. Discovery of Antibiotic (E)-3-(3-Carboxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one. Journal of the American Chemical Society, v. 137, n. 5, p. 1738-1741, 2015. DOI: 10.1021/jacs.5b00056.
COHEN, N. C.; J. M. BLANEY; C. HUMBLET; P. GUND; D. C. BANY. Molecular Modeling Software and Methods for Medicinal Chemistry. Journal of Medicinal Chemistry, v. 33, p. 883-894, 1990. DOI: 10.1021/jm00165a001.
FRISCH, M. J.; et al. Gaussian 09 (Gaussian, Inc.), Wallingford CT, 2009.
IMMING, P. Medicinal Chemistry: definitions and objectives, drug activity phases, drug classification systems. In: WERMUTH, C. (Ed.). The practice of Medicinal Chemistry. 3. ed. [s.l.] Elsevier, p. 63–72, 2008. ISBN 9780123741943.
NEGRELLI, M. Análise Conformacional do CIS E TRANS 2- Hidroxicicloexanocarboxilato de Etila. Dissertação de Mestrado. Universidade Estadual de Ponta Grossa, 2011.
RODRIGUES, C. R. Processos modernos no desenvolvimento de fármacos: modelagem molecular. Cadernos Temáticos de Química Nova na Escola, n. 3, 2001.
SANTOS, C. B. R. Desenvolvimento Racional de Fármacos Antimaláricos Derivados da Artemisinina usando Métodos Computacionais SAR e QSAR. Tese de Doutorado, Universidade Federal do Amazonas, Brasil, 2014.
SANTOS, K. L. B. Planejamento molecular, síntese e relação estrutura-atividade de isoxazolonas benzilidênicas com atividade anti-inflamatória. Tese de Doutorado, Universidade Federal do Pará, Belém, 2021.
SILVERSTEIN, R. M.; WEBSTER, F. X.; KIEMLE, D. Spectrometric Identification of Organics Compounds. New Jersey: Jhon Wiley & Sons, 2005
SOUZA, A. A.; FARIAS, R. F. Elementos de Química Quântica. Campinas: Átomo, 2007.
VERLI, H.; BARREIRO, E. J. Um paradigma da química medicinal: a flexibilidade dos ligantes e receptores. Química Nova, v. 28, n. 1, p. 95–102, jan. 2005.
ZUMDAHL, S. S.; ZUMDAHL, S. A. CHEMISTRY. New York: Houighton Mifflin Company, 2007.