Autores
Santos, V.F. (UNIVERSIDADE FEDERAL DO RIO DE JANEIRO) ; Pereira, H.M.G. (UNIVERSIDADE FEDERAL DO RIO DE JANEIRO) ; Machado, S.P. (UNIVERSIDADE FEDERAL DO RIO DE JANEIRO)
Resumo
Dithiocarbamates play an important role in many fields and act as a ligand for
various transition metals due to the coordination ability of the dithio moiety. In
this work, the Density Functional Theory and Time-Dependent Density Functional
Theory were employed for prediction of the structure and electronic absorption
spectra of tris(diethyldithiocarbamate)rhodium(III). As there was no experimental
X-ray structure available, a previous optimized and validated methodology for
other complexes of the same class was used for achieving the goals. The
computational results were in good agreement with the experimental absorption
spectra at the ultraviolet-visible region available in the literature.
Palavras chaves
Dithiocarbamate; Rhodium; DFT
Introdução
Dithiocarbamates (DTC) are one example of a class of monoanionic 1,1-dithiolate
ligands, with also includes xanthates and thioxanthates (HOGARTH, 2012).
Although they look very similar, the DTC occupy a very special niche and has
been studied in different fields, such as industrial applications, pesticides,
fungicides, and chemotherapy (HOGARTH, 2012; JÚNIOR et al., 2012; MANSOURI et
al., 2016). DTC can also be applied as precursors of metal sulphides, analytical
reagents, reprocessing of polymers, antioxidants, and catalysts (MANSOURI et
al., 2021). Some of the dialkyl-substituted DTC salts have interesting
biological effects and are used as antidotes for cadmium intoxication, and as
antibacterial, antifungal and immunoregulatory agents (JIAN et al., 2002;
MANSOURI et al., 2021). As a ligand, the DTC form stable complexes with
transition metal ions, due to the coordination ability of dithio moiety. Various
oxidation states and coordination geometries can be stabilized by the high
electron density on the sulfur atoms (MANSOURI et al., 2021). Due to their
importance and to the lack of information in the literature, calculations
employing the Density Functional Theory (DFT) and the Time-Dependent Density
Functional Theory (TD-DFT) can give a deeper insight into the structural and
spectroscopic properties for their characterization. In addition, it can be
useful for the predictability of new structure-related complexes’ properties for
other applications in the future. Thus, the main goals are to (1) optimize the
structure of tris(diethyldithiocarbamate)rhodium(III) [Rh(DTC)3] using a
previous validated computational methodology for other complexes of the same
class and (2) to predict its electronic absorption spectra by TD-DFT.
Material e métodos
Calculations employing DFT and TD-DFT were performed with Gaussian16 for
prediction of structural and spectroscopic properties of the low spin Rh(DTC)3
complex. Due to the unavailability of an X-ray structure for the studied
complex, two starting geometries were tested with a previous and validated
computational methodology for Co(DTC)3, Ni(DTC)2 and Pd(DTC)2. For Rh(DTC)3
geometry 1, the X-ray structure available in Cambridge Structural Database (CSD)
for Co(DTC)3 (CSD refcode: ETDCCO02) (HEALY et al., 1990) was used, in which Co
was changed for Rh. In this case, the bond lengths and angles were the same as
the X-ray structure. For geometry 2, the bond lengths and angles of the DTC
moiety of CSD EMOKAU (MANSOURI et al., 2016) were used in the octahedron
geometry of CSD ETDCCO02. Both geometries were optimized, and frequencies were
calculated in the gas phase and in solvent (chloroform, CPCM) with CAM-B3LYP
functional and GEN (6-31G** for S, C, N and H, LanL2DZ for Rh) basis set. The
electronic absorption spectra of the optimized structures were simulated by TD-
DFT with the same computational methodology described above.
Resultado e discussão
The computational methodology employed in this study was validated
experimentally for Co(DTC)3, Ni(DTC)2 and Pd(DTC)2. As there is no X-ray
structure available for Rh(DTC)3, two geometries were tested as starting points,
which are represented in Figure 1. No negative frequencies were observed and the
energy difference between the two optimized structures were 0.003 kcal/mol (gas
phase) and 0.01 kcal/mol (CPCM, chloroform). The same final structure was
obtained in both cases, with bond lengths of 2.4095 angstroms for Rh – S, 1.7288
angstroms for S – C and 1.3254 angstroms for C – N, as shown in Figure 1. The
simulated electronic absorption spectra by TD-DFT in chloroform showed one band
in visible region (439 nm) and bands in the ultraviolet region in 307 nm, 246 nm
and 237 nm. The results were in agreement with the literature (JORGENSEN, 1962),
in which the experimental bands occur in 423 nm, 282 nm, 252 nm and 226 nm,
respectively. As can be seen by this results, DFT and TD-DFT can be a powerful
tool for the predictability of structures and properties for complexes without
experimental data. It is noteworthy to mention that the methodology in these
cases should be validated previously against similar structures (if available)
and/or supported by literature.
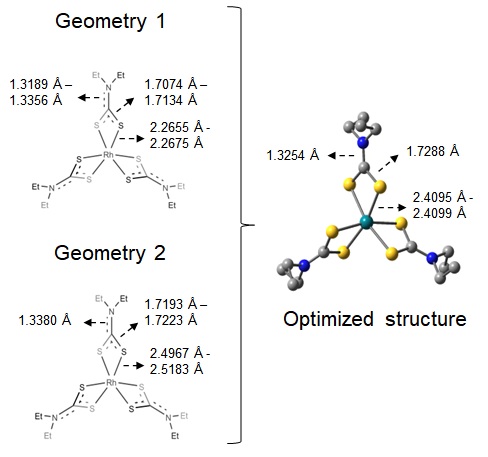
Representation of geometries 1 and 2, and optimized structure for Rh(DTC)3 complex.
Conclusões
As a conclusion, the structural and spectroscopic properties of Rh(DTC)3 were
obtained employing a previous optimized computational methodology for other
complexes of the same class. The CAM-B3LYP functional and GEN (6-31G** for S, C, N
and H, LanL2DZ for Rh) basis set were employed for both geometry optimization and
simulation of electronic absorption spectra. The results are in good agreement
with the literature, showing the potentiality of DFT and TD-DFT for predicting new
structure and properties of inorganic complexes.
Agradecimentos
The authors thank CNPq, FAPERJ and Autoridade Brasileira de Controle de Dopagem
(ABCD) for financial support.
Referências
[1] HEALY, P. C.; CONNOR, J. W.; SKELTON, B. W.; WHITE, A. H. Alkyl substituent effects in diamagnetic dithiocarbamate cobalt (III) and nickel (II) complexes. Australian Journal of Chemistry, no 43, 1083-1095, 1990. [2] HOGARTH, G. Metal-dithiocarbamate complexes: chemistry and biological activity. Mini reviews in medicinal chemistry, no 12, 1202-1215, 2012. [3] JIAN, F., BEI, F., ZHAO, P., WANG, X., FUN, H., CHINNAKALI, K. Synthesis, crystal structure and stability studies of dithiocarbamate complexes of some transition elements (M= Co, Ni, Pd). Journal of Coordination Chemistry, no 55, 429-437, 2002. [4] JORGENSEN, C. K. Absorption spectra of transition group complexes of sulphur-containing ligands. Journal of Inorganic and Nuclear Chemistry, no. 24, 1571-1585, 1962. [5] JÚNIOR, A. C., VERSIANE, O., ONDAR, G. F., RAMOS, J. M., FERREIRA, G. B., MARTIN, A. A., SOTO, C. T. An experimental and theoretical approach of spectroscopic and structural properties of the bis (diethyldithiocarbamate)–cobalt (II). Journal of Molecular Structure, no 1029, 119-134, 2012. [6] MANSOURI, G., HEIDARIZADI, F., NAGHIPOUR, A., NOTASH, B. Synthesis, characterization and antibacterial study of cyclometalated rhodium (III) complex containing dithiocarbamate. Journal of Molecular Structure, no 1121, 128-134, 2016. [7] MANSOURI, G., GHOBADI, M., NOTASH, B. Synthesis, spectroscopic, structural, DFT and antibacterial studies of cyclometalated rhodium (III) complex based on morpholinedithiocarbamate ligand. Inorganic Chemistry Communications, no 130, 108707, 2021.














