Autores
Santos, H.B.T. (UNIVERSIDADE FEDERAL DO PARÁ) ; Araújo, J.O. (UNIVERSIDADE FEDERAL DO PARÁ) ; Marinho, C.V.S. (UNIVERSIDADE FEDERAL DO PARÁ) ; Lima, A.H.L. (UNIVERSIDADE FEDERAL DO PARÁ)
Resumo
A enzima PcEST, do fungo Penicillium chrysogenum, apresenta um enorme potencial
na indústria farmacêutica, pois atua na conversão da catálise da lovastatina em
monacolina J, precursora na produção de sinvastatina, droga redutora do
colesterol. Este trabalho tem como objetivo avaliar a estabilidade da enzima
PcEST mutantes, através de Métodos de dinâmica molecular. Os resultados
mostraram que a mutação D106A apresentou um complexo proteína-ligante com maior
afinidade, o que está de acordo com os resultados experimentais. Ademais a
dinâmica molecular demonstrou que a mutação D106A é mais ativa devido as muitas
interações efetivas. Entretanto a mutação W344K, exibiu a perda de interações
durante a dinâmica molecular com os principais resíduos localizados no sítio
ativo da enzima PcEST.
Palavras chaves
Dinâmica molecular; PcEST; Sinvastatina
Introdução
As doenças cardiovasculares (DCV) configuram uma das principais causas de morte
no mundo e de acordo com a Organização Mundial da Saúde (OMS) 17,9 milhões de
pessoas morrem a cada ano, sendo aproximadamente 32% de todas as mortes no mundo
(WHO, 2022). Dentre os principais fatores de risco para a doença cardiovascular
que podemos destacar está a hipercolesterolemia, doença responsável por elevar
as taxas de colesterol no sangue. O colesterol é um precursor dos hormônios
esteróides e um elemento essencial da membrana celular, contudo, a regulação
alterada da síntese, absorção e excreção do colesterol predispõe a doenças
cardiovasculares de origem aterosclerótica (ZARATE et al., 2016).
Em uma das formas de se combater esta enfermidade, utiliza-se a sinvastatina
sigla IUPAC, [(1S,3R,7S,8S,8aR)-8-[2-[(2R,4R)-4-hydroxy-6oxooan-2-yl]ethyl]-3,7-
dimethyl-1,2,3,7,8,8a-hexahydronaphthalen-1-yl] 2,2-dimethylbutanolenate (Figura
1), fármaco semissintético fabricado através da lovastatina, metabólito
secundário produzido pelo fungo Aspergillius terreus. A sinvastatina pertence à
classe de fármacos inibidores da hidroximetilglutaril-coenzimaA (HMG-CoA)
reductase, este medicamento tem como função atuar na redução significativa do
mau colesterol, LDL (Low Density Lipoprotein), bem como dos triglicérides
(substância gordurosa). A sinvastatina também aumenta os níveis do bom
colesterol, HDL (High Density Lipoprotein), no sangue, reduzindo os níveis de
doenças cardíacas relacionadas à hipercolesterolemia (XIE; TANG, 2007).
Entretanto, seu processo de produção é complexo, trabalhoso, caro e prejudicial
ao meio ambiente, pois são utilizados uma alta concentração de reagentes ácidos,
alcalinos e orgânicos em sua formação. Desta forma, a biossíntese enzimática
utilizando uma hidrolase específica de lovastatina é uma das formas alternativas
para a produção sustentável de sinvastatina (LIANG; LU., 2020).
Neste âmbito a química medicinal se propõe a estudar as razões moleculares da
ação dos fármacos, a relação estrutura química e a atividade farmacológica,
incluindo o planejamento e o desenho estrutural de novas substâncias que possuam
propriedades farmacoterapêuticas úteis capazes de representarem novos fármacos
(BARREIRO; FRAGA, 2021). Desta forma, o desenvolvimento de enzimas na
biossíntese de sinvastatina tem auxiliado na descoberta de novos candidatos a
fármacos.
Deste modo, a PcEST, enzima do fungo Penicillium chrysogenum, apresenta um
enorme potencial na indústria farmacêutica, haja vista que possui a função de
hidrolisar a cadeia lateral da lovastatina para produzir monacolina J, principal
precursor para a síntese de sinvastatina, fármaco importante para o tratamento
da hipercolesterolemia (XIE; TANG, 2007).
Contudo, estudos relatam que a enzima PcEST apresenta baixa solubilidade e
termoestabilidade (LIANG et al, 2018). As posições de alta flexibilidade das
proteínas podem afetar a solubilidade e/ou diminuem sua termoestabilidade, uma
vez que elevam a entropia durante o desdobramento de proteínas, aumentado a
quantidade de conformações desdobradas (FANG et al., 2014; LIANG; LU., 2020;
MALAKAUSKAS; MAYO., 1998; YAINOY et al., 2019).
Nesta perspectiva, este trabalho tem como objetivo simular a enzima PcEST,
através da avaliação de estabilidade, por meio dos métodos computacionais de
dinâmica molecular, em busca de entender a atividade catalítica da enzima PcEST,
deste modo agregando informações para aumentar a sua resistência nessas
condições e contribuir para a produção de Monacolina J e consequentemente para a
síntese de sinvastatina. As informações obtidas a partir de sua análise serão de
suma importância para a conjectura de novas variantes da enzima PcEST propostas
para futura síntese e ensaios biológicos
Material e métodos
Inicialmente, após a busca de dados na literatura foi obtida a estrutura
tridimensional da enzima PcEST do fungo Penicillium chrysogenum, pelo banco de
dados de proteína (PDB) com o código 6KJD (LIANG; LU, 2020). Para a preparação
dos sistemas utilizou-se a enzima PcEST em complexo com o substrato Lovastatina.
As mutações constituíram na produção de sete mutações, são elas: D106A, D131A,
S57A, W344F, W344K, Y127A, Y127F, conforme a literatura.
Para o preparo das estruturas utilizou-se o servidor PDB2PQR. Os estados de
protonação foram atribuídos pelo programa PROPKA à pH 7.0. Para o preparo do
substrato utilizou-se a lovastatina presente na estrutura cristalográfica com o
código pdb (6KJD), as cargas da lovastatina foram calculadas no programa
Gaussian 09, utilizando o método Hartree-Fock com o conjunto de base 6-31G*.
Com o intuito de investigar a estabilidade da enzima PcEST realizou-se a
simulação de dinâmica molecular utilizando o programa Amber18 (CASE et al,
2005). Utilizou-se o módulo tLeap para criar os arquivos de entrada para a
simulação de DM, o campo de força Amber ff14SB (KAMENIK et al., 2020) foi
utilizado para descrever o conjunto de átomos da enzima. Em seguida, o sistema
foi solvatado em uma caixa de formato cúbico representada pelo modelo de água
TIP3P com tamanho 12 Å entre o limite da caixa e os átomos do sistema usando
condições periódicas de contorno. Íons de Na+ foram incluídos no intuito de
preservar a eletroneutralidade do sistema.
A Dinâmica molecular envolve quatro etapas: minimização de energia, aquecimento,
equilíbrio e simulação de dinâmica de produção (YE; LI; HU, 2021). Primeiramente
todas as moléculas de água e contra íons foram minimizadas por 8000 ciclos, logo
após todos átomos de hidrogênio da enzima foram minimizados por 3000 ciclos
utilizando o método steepest-descent seguido de 2000 ciclos de gradiente
conjugados, os átomos de hidrogênio das moléculas de água foram minimizados por
8000 ciclos, por último todo sistema foi minimizado por 5000 ciclos com
steepest-descente e 5000 ciclos de gradiente conjugado totalizando 10000 ciclos.
Após a minimização, iniciou-se o aquecimento do sistema utilizando o termostato
de Langevin, através de 200 picossegundos de dinâmica molecular com o objetivo
de aquecer o sistema gradativamente de 0 a 303,15 K (30 ºC). Todos os sistemas
foram equilibrados para iniciar a etapa da produção de 500 ns de DM com ensemble
NPT à pressão de (1atm) e temperatura de (303,15 K) constantes. Para diferenciar
as interações de longa distância utilizou-se o raio de corte (cutoff) de 8, 0 Å.
O algoritmo SHAKE (SONG et al., 2019) foi empregado para restringir as ligações
envolvendo átomos de hidrogênio mantendo-os em suas distâncias de equilíbrio
pré-definidas durante as minimizações possibilitando utilizar um passo de
integração de 2 fs para realização dos cálculos de DM.
Resultado e discussão
A amostragem foi obtida para os sistemas mutantes para avaliar parâmetros
estruturais e energéticos dos sistemas de estudo. No estudo acerca da enzima
PcEST, a presença das mutações (D106A, D131A, S57A, W344F, W344K, Y127A e Y127F)
podem desempenhar uma função estrutural relevante em relação a estabilidade e
dinâmica.
As evoluções das conformações nos sistemas foram analisadas através do desvio
médio quadrático (do inglês, Root Mean Square Deviation − RMSD), bem como da
flutuação quadrática média (do inglês, Root Mean Square Fluctuation −RMSF) de
cada estrutura com relação à estrutura de referência do passo de equilíbrio,
calculado após o alinhamento baseado nos átomos da espinha dorsal.
Figura 1, mostra o gráfico RMSD, em que o receptor é representado na cor preto,
o complexo pela cor vermelha e os ligantes representados por suas respectivas
cores. Por meio da análise o gráfico RMSD exibe uma variação estável nos
sistemas D106A, D131A, S57A, W344F, Y127A e Y127F, onde os valores RMSD de todos
os complexos encontravam-se dentro de uma flutuação aceitável em uma faixa de 1
a 3Å indicando o alcance do equilíbrio estrutural destes sistemas.
Conquanto, o sistema W344K apresentou uma elevada variação, de acordo o gráfico
RMSD, com variação de 1 à 10 Å, apontando a instabilidade neste sistema ao longo
da simulação de dinâmica molecular, além disso a mutação Y127A sofreu uma
elevada oscilação ao longo da DM, no entanto permaneceu estável dentro dos 3 Å.
Deste modo, a partir da visualização das estruturas apresentadas nas trajetórias
é possível notar um elevado movimento do complexo com a mutação W344K, quando
comparado as demais mutações, que apresentaram oscilações moderadas.
Os resultados alcançados com o gráfico RMSF apontam sentidos semelhantes entre
todos os sistemas. Os resíduos que possuem maiores valores de flutuação se
encontram na região C-terminal Através do estudo é possível observar que as
flutuações relevantes encontram-se presentes na região de loop R2 (127-153), com
oscilação que se aproxima à 5 Å, e loop R1 (222-248), com flutuações próximas a
4 Å. As regiões N-terminais formam diferentes oscilações, enquanto as regiões C-
terminais formaram flutuações semelhantes com flutuação que chega próximo à 6 Å.
Observou-se que os ligantes apresentaram interações de hidrogênio diferente uma
das outras. Ademais, conforme os resultados obtidos, as interações de hidrogênio
mais estáveis presentes no sítio catalítico da proteína envolvem os resíduos
presentes no Loop R1 das enzimas mutantes. Estudos apontam a importância dos
loops R1 (resíduos 222-248), R2 (resíduos 127-153) e R3 (resíduos 290-319) ao
redor do sítio catalítico, pois são capazes de determinar a abertura e o fundo
da bolsa de ligação ao substrato, respectivamente, atuando como uma trava para
abertura do túnel de ligação do ligante (LIANG; LU., 2020).
Por meio das análises realizadas, verificou-se que todos os sistemas apresentam
divergências em suas interações de hidrogênio, em virtude de suas mutações. A
mutação D106A formou interação com o resíduo Glu242, o mutante D131A apresentou
interação com o resíduo Ser58, já o mutante S57A, formou interação de hidrogênio
com o resíduo Gly348. Os mutantes W344F e W344K não apresentaram interações de
hidrogênio significativas. A mutação Y127A, apresentou uma ligação de hidrogênio
com o resíduo Arg139. O mutante Y127F apresentou interações com os resíduos
Ser58 e Tyr171. Ademais, verificou-se que os resíduos Gly348, Phe130 e Phe310
contribuem na estabilidade do ligante lovastatina devido à formação de
interações de van der Waals.
Ademais, foi realizado um estudo de análise energética, nesta análise, utilizou-
se os últimos 100ns de simulação de Dinâmica Molecular dos sistemas mutantes
para os cálculos de energia livre de ligação proteína-ligante usando o método
SIE . Na análise energética utiliza-se uma parte da trajetória em que os
intervalos estabelecidos (últimos 100ns) foram selecionados. Sendo utilizados
10.000 frames com intervalo igual a 2, resultando em 5.000 frames para o
cálculo.
O método utilizado foi capaz de predizer a afinidade de ligação de todos os
sistemas. Empregando a abordagem SIE, foi possível obter os seguintes valores de
energia, -8,48 kcal/mol para o mutante D106A, com melhor afinidade e -2.82
kcal/mol para o mutante W344K de menor afinidade.
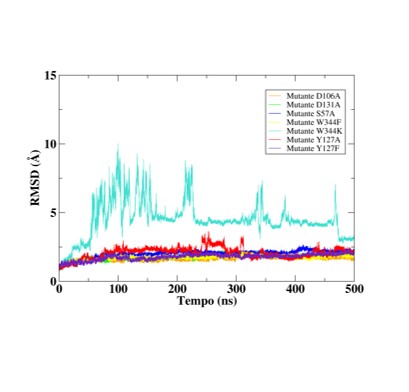
Representação do gráfico RMSD da simulação de Dinâmica Molecular.
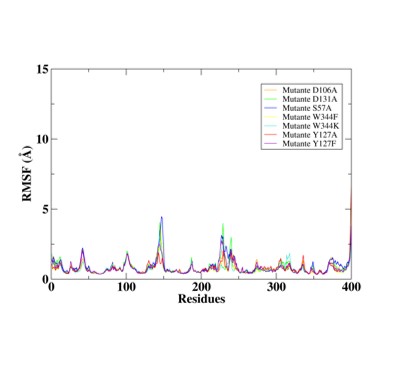
Representação do Gráfico RMSF ao longo da simulação de dinâmica molecular.
Conclusões
Neste estudo, empregamos a dinâmica molecular e cálculos de energia livre de
ligação para fornecer insights sobre s estabilidade da PcEST, Enzima responsável
pela hidrólise da lovastatina em Monacolina J, principal precursora na síntese
de sinvastatina. De acordo com os nossos resultados a mutação D106A apresentou o
complexo proteína-ligante (∆G bind) com maior afinidade o que está de acordo com
os resultados experimentais que o representam com maior estabilidade em termos
de termoestabilidade e solubilidade. Entretanto, a mutação W344K, exibiu menor
afinidade devido o rompimento das interações de hidrogênio, bem como a perda
interações durante a simulação de dinâmica molecular com os principais resíduos
localizados no sítio ativo da enzima PcEST
Nossos resultados apresentaram que essa alta afinidade deve-se à relativa alta
no número de interações com os resíduos-chave nos sítios ativos da enzima, em
especial Ser58, Phe130, Tyr171, Phe310, Trp345 e Gly348. Além disso os resíduos
Phe 130 e Phe310 formam empilhamentos pi com o anel naftaleno da lovastatina,
apresentando forte interações de van der Waals. Portanto, este estudo permite
uma interpretação molecular das principais interações que ocorrem entre a enzima
PcEST e seu substrato lovastatina, contribuindo assim para uma melhor análise e
compreensão dos processos catalíticos desse alvo biológico e auxiliando no
processo de novos candidatos a fármacos. Espera-se que os resultados aqui
relatados possam ser úteis para agregar informações para aumentar a sua
resistência nessas condições e contribuir para a produção de Monacolina J e
consequentemente para a síntese de sinvastatina. Deste modo, as informações
obtidas a partir de sua análise serão de suma importância para a conjectura de
novas variantes da enzima PcEST propostas para futura síntese e ensaios
biológicos.
Agradecimentos
Agradeço à toda equipe LPDF (laboratório de planejamento e desenvolvimento de
fármacos) pelo suporte e ensinamentos, ao programa PPGQMMM, à Fapespa pela bolsa
de pesquisa e a meus familiares e amigos pelo apoio e incentivo.
Referências
BARREIRO, Elizer J; FRAGA, Carlos Alberto Manssour. Química medicinal: as bases moleculares da ação dos fármacos. Porto Alegre. Artmed Editora, 2001.
CASE, D. A. et al. The Amber biomolecular simulation programs. Journal of computational chemistry. v. 26, n. 16, p. 1668–1688, dez. 2005.
FANG Z, et al. Structure-based rational design to enhance the solubility and thermostability of a bacterial laccase Lac15. PLoS One. V.9, ed. 7, 18 Jul 2014.
KARPLUS M; MCCAMMON JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol. v. 9, ed. 9, p. 646652, 01 Sep. 2002.
KAMENIK, Anna S. et al. Polarizable and non-polarizable force fields: protein folding, unfolding, and misfolding. The Journal of Chemical Physics. v. 153, ed. 18, p. 185102, 13 Nov. 2020.
LIU, Xuewei. et al. Molecular dynamics simulations and novel drug discovery. Expert Opin Drug Discov. v. 13, ed. 1, p. 2337, Jan. 2018.
LIANG, Y; LU, X. Structural insights into the catalytic mechanism of lovastatin hydrolase. Journal of Biological Chemistry. v. 295, ed. 4, p. 10471055, Jan. 2020.
MALAKAUSKAS SM, MAYO SL. Design, structure and stability of a hyperthermophilic protein variant. Nat Struct Biol. ed 5, p 470-475, Jun 1998. doi: 10.1038/nsb0698-470.
SONG, Lin Frank. et al. Using AMBER18 for Relative Free Energy Calculations. J. Chem Inf. Model. v. 59, ed. 7, p. 31283135, 22 Jul. 2019.
XIE, X; TANG Y. Efficient synthesis of simvastatin by use of whole-cell biocatalysis. Appl Environ Microbiol. v. 73, ed. 7, p. 20542060. 01 Apr. 2007.
YE J; LI, L; HU, Z. Exploring the Molecular Mechanism of Action of Yinchen Wuling Powder for the Treatment of Hyperlipidemia, Using Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation. Biomed Res Int. v. 2021, 28 Oct. 2021.
ZÁRATE A, et al. Cholesterol and atherosclerosis. Historical considerations and treatment. Arch Cardiol Mex. v. 2, p. 163-169. 7 Jan 2016.














