Autores
Marinho, C.V.S. (UFPA) ; Santos, A.M. (UNICAMP) ; Araújo, J.O. (UFPA) ; Santos, H.B.T. (UFPA) ; Lima, A.D.N. (UFPA) ; Lima, A.H.L. (UFPA)
Resumo
A estabilidade entre os complexos de
proteínas Mur é essencial para se conhecer
as possíveis conformações contínuas que os
heterodimeros podem assumir em meio
biológico. Foram estudadas por meio de
Ancoragem e Dinâmica molecular as
conformacões adotadas para cada dímero
formado pelas Mur ligases sendo eles
MurC:MurF e MurD:MurF. A ancoragem
molecular foi feita com base em diferentes
servidores e algoritmos para a formação de
cada dimero. A dinâmica molecular analisou
a estabilidade em 50ns de simulação,
separando os melhores servidores e
conformações correspondentes. Concluímos
que o servidor AlphaFold foi o melhor para
o dimero MurC:MurF e o servidor Gramm-X
foi o melhor para o dimero MurD:MurF. Com
isso separa-se que seja possível a
determinação futura do dímero nativo das
Mur ligases.
Palavras chaves
Dinâmica Molecular; Complexos Binários; Mur Ligases
Introdução
O Streptococcus pneumoniae é o
principal
organismo causador da doença
“pneumonia”
adquirida entre crianças e adultos
no
mundo, causando milhões de mortes
por ano
e contando com um custo financeiro
alto
para o controle da doença (MICHELIN
et
al., 2019).
As enzimas Mur são responsáveis pela
síntese dos lipídios I e II
presentes no
peptidoglicano (PG) um dos
principais
componentes da parede celular
responsável
pela estrutura e estabilidade da
célula
(LADDOMADA et al., 2016). Neste
trabalho
analisamos a atividade entres as
enzimas
Mur do patógeno humano Streptococcus
pneumoniae R6, com a finalidade de
investigar a estabilidade estrutural
entre
os complexos formados pelas
enzimas
MurC:MurF e MurD:MurF (MIYACHIRO et
al.,
2019). A estabilidade entre as
proteínas
Mur é essencial para se conhecer as
possíveis conformações contínuas que
o
heterodimero estudado pode assumir
em meio
biológico.
Estudos utilizando métodos de
química
computacional como a homologia de
estruturas, a docagem molecular e
simulações de Dinâmica Molecular
(DM) têm
se mostrado promissores na
prospecção de
compostos multi-alvos na biossíntese
de PG
(KUMARI; SUBBARAO, 2021), espera-se
que os
resultados possam contribuir para o
planejamento racional de fármacos
multi-
alvos e ratificar a importância e
potencial da polifarmacologia,
principalmente no tratamento de
doenças
bacterianas (BARREIRO et al., 1997).
Material e métodos
A homologia das estruturas foi
feita
após a obtenção da sequência de
aminoácidos (FASTA) de cada enzima
depositadas no banco de dados
Protein
Data Bank e foi realizado o
alinhamento
entre o organismo existente a cada
enzima
e o organismo alvo no servidor BLAST
que
fez alinhamento a partir da
comparação de
dados selecionados resultando em
sequências FASTA ranqueadas de
acordo com
a similaridade com o organismo alvo.
A
modelagem das proteínas foi feita no
Swiss-Model (WATERHOUSE et al.,2018)
que a
partir da FASTA submetida realiza a
modelagem comparativa gerando os
modelos
de cada proteína e o Alpha Fold que
combina as informações presentes da
sequência de aminoácido das
proteínas
para prever a estruturas das cadeias
(EVANS et al. 2021).
Para a realização da docagem
molecular
foram utilizados diferentes
servidores
online: Haddock no qual agrega sete
interfaces diferentes associada a um
nível
de controle sobre o protocolo de
encaixe
refletido (VAN ZUNDERT et al. 2016),
FireDock que utiliza um método de
refinamento das proteínas e as
classifica
de acordo com a energia livre de
ligação
de cada uma (MASHIACH et al. 2008),
Gramm-
X que utiliza a melhor
correspondência
entre a superfície das enzimas
usando a
correlação pelo método de FFT
(TOVCHIGRECHKO AND VAKSER. 2006),
MdockPP
que faz uso de uma previsão
hierárquica
estrutural das proteínas também por
meio
de método FFT e faz uma Avaliação
Crítica
de Predição de Interações (XU et al.
2016).
A DM foi realizada em etapas, uma na
qual
descartamos os complexos menos
estáveis
fazendo uma simulação de 20ns. Em
seguida,
continuou a simulação partindo dos
20ns
até 50ns para a análise da
manutenção do
número das ligações de hidrogênio
(JANDOVA
et al. 2021). O pacote AMBER foi
utilizado
para o preparo e a execução das
simulações
(CASE et al. 2005). O campo de força
ff14SB foi utilizado para construir
a
topologia e os parâmetros dos
sistemas
proteicos. O módulo tLeap foi usado
para
solvatar o sistema em uma caixa
cúbica com
água explícita do tipo TIP3P.
Foram realizadas 4 etapas de
minimização:
a primeira minimizando a água e os
contra
íons em um total de 8000 ciclos, a
segunda
minimizando o complexo proteico em
um
total de 5000 ciclos, a terceira
minimizando o complexo proteico e a
água
em um total de 4000 ciclos e a
quarta
minimiza todos os componentes em um
total
de 10000 ciclos. Em seguida, foi
realizada
a etapa de aquecimento dos sistemas
gradualmente de 0 a 298 K utilizando
o
termostato de Langevin para o
controle de
temperatura com um tempo de
simulação a
200 ps, continuando com a
equilibração
mantendo a temperatura constante em
298 K
utilizando o barostato Berendsen.
Resultado e discussão
A Construção dos modelos foi feita a partir do Swiss-model que gerou estruturas
tridimensionais de cada proteína modelada, essas estruturas foram validadas a partir
da análise do gráfico Ramachandran que mostrou a porcentagem de configurações prováveis de 94,01% para MurF, 92,38% para MurC e 97,38% para MurD. O Alpha Fold foi usado para a construção dos complexos direto pela sequência obtida no BLAST sem a necessidade de uma docagem posterior, e a sua validação mostrou 96.80% de configurações prováveis para os dois
complexos.
Para os complexos MurC:MurF e MurD:MurF, a melhor docagem em questão de alinhamento
em comparação foi à do servidor Haddock. Os complexos resultante da docagem molecular foram comparados com a ferramenta "Matchmaker" que alinha as poses carregadas no programa UCSF Chimera e foi obtido um resultado inesperado pois o alinhamento de todos os servidores juntos cada pose localizou-se em um local diferente da proteína, mostrando que mesmo com parâmetros diferentes não houve poses em domínios iguais, assim análise seguiu para classificar qual servidor gerou o complexo que seria o mais estável, através de uma análise da estabilidade de cada um a partir da DM.
A produção de 20 ns de DM foi analisada com o cálculo de RMSD (do inglês: Root Mean Square Deviation) para cada sistema, que calcula o desvio dos átomos dos complexos com base em uma estrutura de referência, a partir desse cálculo foram gerados gráficos que auxiliaram a avaliação da estabilidade de cada complexo proveniente da docagem molecular depois da DM.
Desde a primeira fase da DM percebeu-se que os complexos com o maior número de ligações de hidrogênio (LH) entre elas as proteínas eram aqueles com maior estabilidade. O servidor FireDock
obteve uma instabilidade muito grande para os dois complexos: MurC:MurF e MurD:MurF, assim ele foi descartado devido a sua elevada movimentação durante as simulações e não obter LH entre o complexo significativa.
Para o complexo MurC:MurF a primeira fase nos mostrou que os servidores MDock, AlphaFold e Gramm-X eram os mais estáveis. O servidor Haddock se mostrou instável no início da simulação, no entanto obteve com uma pequena estabilidade no final, para avaliar de fato essa possível estabilidade foi necessário a continuação da simulação, passando-o assim para segunda fase de DM. Na segunda simulação os servidores Alpha Fold, Haddock e MDock foram os melhores complexos pois mantiveram e aumentaram as LH entre as proteínas assim aumentando sua estabilidade.
Para um melhor refinamento do melhor servidor, foi comparado entre três melhores resultados o desvio padrão das distâncias dos átomos obtido a partir do gráfico RMSD, assim o melhor resultado seria aquele com o menor valor do desvio padrão. O complexo de MDock obteve 1,01 σ de desvio padrão e foi o mais elevado em comparação aos outros dois que obtiveram um valor de desvio padrão semelhantes para o Haddock um valor de 0,69 σ e para o AlphaFold um valor de 0,51 σ. Nos servidores Haddock e AlphaFold foram os mais estáveis após a finalização da produção de DM.
Para o complexo MurD:MurF a primeira simulação de DM mostrou que dois servidores obtiveram uma instabilidade muito grande: Alpha Fold e FireDock. O resultado do Alpha Fold foi surpreendente, pois foi o servidor mais instável da DM, sem nenhuma LH entre as proteínas ao fim da simulação. Os servidores Mdock, Gramm-x e Haddock foram os melhores respectivamente nesta fase da DM. Na segunda fase de simulação obtivemos o MDockPP como melhor resultado justamente por ser o servidor mais estável e com um desvio de 0,28 σ seguido apenas pelo Gramm-X que resultou em um desvio de 0,41 σ iniciando a simulação com uma estabilidade aceitável e terminando com um pico de instabilidade relativamente grande.

a estabilidade dos servidores a partir da dinâmica molecular para o complexo MurD e MurF.
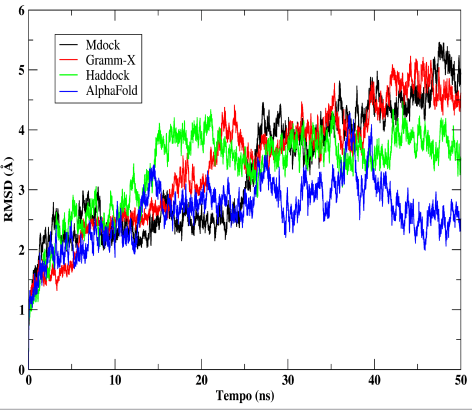
A estabilidade dos servidores a partir da dinâmica molecular para o complexo MurC e MurF.
Conclusões
Os resultados refinaram a busca pela
conformação mais estável dos heterodimeros
formados pelas Mur ligases. A ancoragem
molecular nos mostrou as possíveis
conformações estruturais que cada dímero
poderia assumir, resultando em várias
poses com certa semelhança para cada
dimero, vale ressalta que para o complexo
MurC e MurF as poses ficaram uma em cada
domínio da proteína diferente do
heterodimero MurD e MurF que somente um
complexo se posicionou em um domínio
proteico diferente. A dinâmica molecular
avaliou o início da estabilidade de cada
dímero resultando em 2 conformações
possivelmente estáveis para cada sistema
Agradecimentos
Agradeço a toda a equipe do LPDF (
laboratório de planejamento e
desenvolvimento de fármacos) pela ajuda e
ensinamentos, ao programa de iniciação
científica da UFPA pela bolsa de pesquisa e os meus amigos e família pelo apoio.
Referências
BARREIRO, ELIEZER J., “Modelagem Molecular: Uma Ferramenta Para o Planejamento Racional De Fármacos Em Química medicinal.” Química Nova, vol.20, no. 3, june 1997. pp. 300-310,10.1590/s0100-40421997000300011;
CASE, D. A. et al. The Amber biomolecular simulation programs. Journal of Computational Chemistry, v. 26, n. 16, p. 1668–1688, 2005;
EVANS, RICHARD, et al. Protein Complex Prediction with AlphaFold-Multimer. 4 Oct. 2021, 10.1101/2021.10.04.463034;
JANDOVA, ZUZANA, et al. “Native or Non-Native Protein–Protein Docking Models? Molecular Dynamics to the Rescue.” Journal of Chemical Theory and Computation, vol. 17, no. 9, 3 Aug. 2021, pp. 5944–5954,10.1021/acs.jctc.1c00336;
KUMARI, MADHULATA, AND NAIDU SUBBARAO. “Deep Learning Model for
Virtual Screening of Novel 3C-like Protease Enzyme Inhibitors Against
SARS Coronavirus Diseases.” Computers in Biology and Medicine, vol. 132,
no. 1,May 2021, p. 104317, 10.1016/j.compbiomed.2021.104317. Accessed 18
Apr. 2021;
LADDOMADA, FEDERICA, et al. “Structural Insights into Protein-Protein Interactions Involved in Bacterial Cell Wall Biogenesis.” Antibiotics, vol. 5, no. 2, 28 Apr. 2016, p. 14, 10.3390/antibiotics5020014;
MICHELIN, L. et al. “Mortalidade e custos da pneumonia pneumocócica em adultos: um estudo transversal”. Jornal Brasileiro de Pneumologia, v. 45, 17 out. 2019;
MIYACHIRO, MAYARA M., et al. “Complex Formation between Mur
Enzymes from Streptococcus Pneumoniae.” Biochemistry, vol. 58, no. 30, 2
July 2019,pp. 3314–3324, 10.1021/acs.biochem.9b00277;
MASHIACH, EFRAT, et al. “An Integrated Suite of Fast Docking Algorithms.” Proteins: Structure, Function, and Bioinformatics, vol. 78, no. 15, 6 July 2010, pp. 3197–3204, 10.1002/prot.22790;
TOVCHIGRECHKO, A., AND I. A. VAKSER. “GRAMM-X Public Web Server for Protein-Protein Docking.” Nucleic Acids Research, vol. 34, no. Web Server, 1 July 2006, pp. W310–W314, 10.1093/nar/gkl206;
VAN ZUNDERT, G.C.P., et al. “The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes.” Journal of Molecular Biology, vol. 428, no. 4, Feb. 2016, pp. 720–725, 10.1016/j.jmb.2015.09.014;
WATERHOUSE, ANDREW, et al. “SWISS-MODEL: Homology Modelling of Protein Structures and Complexes.” Nucleic Acids Research, vol. 46, no. W1, 21 May 2018, pp.W296–W303, 10.1093/nar/gky427;
XU, XIANJIN, et al. “Performance of MDockPP in CAPRI Rounds 28-29 and 31-35 Including the Prediction of Water-Mediated Interactions.” Proteins: Structure, Function, and Bioinformatics, vol. 85, no. 3, 2 Dec. 2016, pp. 424–434, 10.1002/prot.25203.














