Autores
Castro Silva Junior, H. (UNIVERSIDADE FEDERAL DO RIO GRANDE DO SUL) ; Silva Menzes, H.N. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Braga Ferreira, G. (UNIVERSIDADE FEDERAL FLUMINENSE)
Resumo
Cálculos SA-CASSCF/NEVPT2 e DLPNO-CCSD(T) foram empregados no estudo da estrutura
eletrônica de quatro complexos de elementos representativos do ligante dmit para
investigar a razão por trás da atribuição de transições eletrônicas próximas à
degenerescência obtidas no espectro UV-Vis reportado na literatura. O estudo
computacional por métodos ab initio mostrou que a forte participação de
correlação estática e dinâmica nos estados excitados dessas moléculas foi oriunda
de sua simetria que justificou a quasi-degenerescência observada.
Palavras chaves
Complexos de dmit; Cálculos ab initio; Estados excitados
Introdução
Complexos do ligante 1,3-ditiola-2-tiona-4,5-ditiolato (dmit) e seus análogos
são estudados há mais de 40 anos, especialmente devido as suas propriedades
eletrônicas que permitem aplicações em supercondutores, semicondutores e metais
moleculares por conta de, entre outras características, orbitais deslocalizados
(DE CARO et al., 2014; GIVAJA et al., 2012; KATO et al., 2017; VELHO; SILVA;
BELO, 2021). No caso de centros catiônicos paramagnéticos, existe ainda a
possibilidade de uso na forma de magnetos moleculares (ATZORI et al., 2016).
Em estudos computacionais publicados na literatura dedicados à investigação da
estrutura eletrônica de complexos dmit, pode ser observado que os espectros no
ultravioleta-visível (UV-Vis), apesar da concordância entre as bandas
experimentais e calculadas, apresentam transições eletrônicas com valores de
energia bastante próximos entre si (quasi-degenerescência) e que não podem ser
explicados à luz da teoria do funcional de densidade que foi o principal método
empregado nestes trabalhos(FERREIRA, 2007; FERREIRA et al., 2008; PAES et al.,
2019). A presença de (quasi)degenerescência já foi reportada em ligantes
análogos ao dmit como o ditioleno (KIRK; MCNAUGHTON; HELTON, 2004).
As características observadas podem ser atribuídas a uma possível natureza
multirreferencial e/ou multiconfiguracional, dado que simetria pode provocar
esse tipo de característica em moléculas. Essa hipótese motivou o emprego de
técnicas ab initio de alto nível, já que estes métodos permitem a construção de
funções de onda com características multirreferenciais e/ou
multiconfiguracionais (ROOS et al., 2016). Neste trabalho, será feito o estudo
da estrutura eletrônica de complexos bi-coordenados de Bi(III) e Sb(III) e
tricoordenados Sn(IV) e Sb(V) com o ligante dmit.
Material e métodos
A primeira etapa foi a otimização geométrica das estruturas. Este procedimento
foi feito por DFT com o método PBEh-3c, que é destinado a otimizações
geométricas(GRIMME et al., 2015). A segunda etapa foi a realização da análise
Fractional Occupation Number Weighted Density (FOD), também via DFT, com o
funcional TPSS que é o mais comum para este cálculo, destinado a facilitar a
compreensão química dos orbitais moleculares por apontarem a localização de
possível correlação eletrônica (BAUER; HANSEN; GRIMME, 2017). A terceira etapa
foi a realização dos cálculos SA-CASSCF com correção perturbacional
NEVPT2(ANGELI et al., 2001) usando os orbitais gerados na etapa anterior para
alimentar o cálculo e selecionar o espaço ativo. Os espaços ativos usados são:
CAS(8,8) para Bi(III) e Sb(III); CAS(10,9) para Sn(IV); e CAS(12,10) para Sb(V).
A quarta etapa foi a realização de cálculos DLPNO-CCSD(T) para compreender
melhor a correlação eletrônica do sistema. Este cálculo foi feito selecionando o
centro metálico e cada ligante como fragmentos separados a ter as interações
eletrônicas observadas. Os cálculos foram feitos no software ORCA 4.2.1(NEESE,
2018) e ORCA 5.0.3(NEESE, 2022) com funções de base Def2-TZVP nos computadores
do Laboratório Multiusuário de Química Computacional da Universidade Federal
Fluminense (LMQC-UFF) e no supercomputador Lobo Carneiro (LoboC). O complexo de
Bi(III) recebeu correção relativística em suas funções de base pelo zeroth order
regular approximation (ZORA)(PANTAZIS et al., 2008).
Resultado e discussão
Os resultados serão detalhados para o complexo de Bi(III) como exemplo. Na
figura 1, podem ser vistas transições eletrônicas do complexo de Bi(III)
calculadas por vários métodos levando à quasi-degenerescência de estados
(FERREIRA, 2007). Porém, a razão não é elucidada, apenas a hipótese de caráter
multirreferencial e/ou multiconfiguracional é levantada na literatura (PAES et
al., 2019).
Os cálculos SA-CASSCF mostraram orbitais simétricos e quasi-degenerados (figura
2), bem como quasi-degenerescência de estados excitados. A energia desses
estados tem o comportamento reportado por FERREIRA, 2007. A natureza
multirreferencial dos sistemas é dada pela quasi-degenerescência dos estados
excitados e se manifesta apenas nos complexos de Bi(III) e Sb(III) (figura 3).
Os complexos de Sn(IV) e Sb(V) são multiconfiguracionais. Para todos os
compostos, a correção NEVPT2 indica correlação dinâmica fortemente atuante nos
complexos, pois alterou significativamente as transições calculadas.
Apesar do número limitado de orbitais, o SA-CASSCF obteve resultado melhor que
os métodos CIS, CISD e TD-RHF reportados na figura 1 na descrição da energia da
primeira banda do espectro UV-Vis do complexo de Bi(III), caracterizada como
π(Sm)→σ*(Bi-S). Esta transição ocorre experimentalmente em 562 e 529 nm (2,2 e
2,3 eV) (FERREIRA et al, 2008), enquanto o cálculo reporta 2,3 e 2,5 eV (figura
3). Porém, a ordem energética dos orbitais diverge dos resultados de FERREIRA et
al, 2000.
A decomposição energética DLPNO-CCSD(T) mostrou que as interações eletrostática,
troca e correlação se manifestam de forma simétrica para cada par ligante-
ligante e ligante-cátion. Reforçando a simetria observada por SA-CASSCF, com as
interações de correlação como a principal fonte de estabilização.
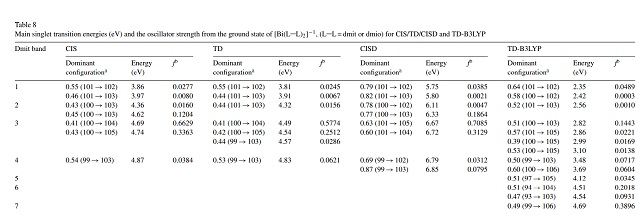
São apresentadas as transições eletrônicas do complexo de Bi(III) cálculadas pot CIS, TD-RHF, CISD e TD-B3LYP retiradas de FERREIRA et al, 2008.
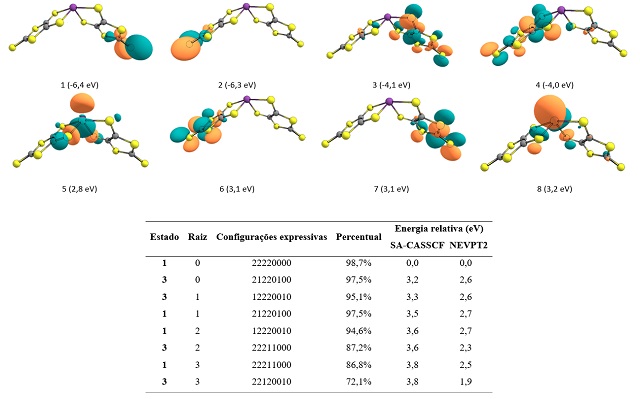
São mostrados os orbitais ativos SA-CASSCF/NEVPT2 (isosuperfície 0,04) do Bi(III) e os dados para os primeiros estados excitados obtidos no cálculo.
Conclusões
O uso do método SA-CASSCF/NEVPT2 confirmou a presença de caráter
multirreferencial nos complexos de Bi(III) e Sb(III) e multiconfiguracional nos
complexos de Sn(IV) e Sb(V) para os estados eletrônicos excitados, o que explica
a tendência a transições quasi-degeneradas anteriormente observadas no espectro
UV-vis. Os cálculos DLPNO-CCSD(T) forneceram uma compreensão detalhada da
dinâmica de interações estabilizantes/desestabilizantes existente entre os
cátions com os ligantes dmit.
Agradecimentos
Os autores agradecem as instituições de fomento PIBIC-UFF e FAPERJ, bem como
ao LMQC-UFF e LoboC-NACAD-UFRJ pelos computadores disponibilizados.
Referências
ANGELI, C. et al. Introduction of n-electron valence states for multireference perturbation theory. Journal of Chemical Physics, v. 114, n. 23, 2001.
ATZORI, M. et al. Quantum Coherence Times Enhancement in Vanadium(IV)-based Potential Molecular Qubits: The Key Role of the Vanadyl Moiety. Journal of the American Chemical Society, v. 138, n. 35, 2016.
BAUER, C. A.; HANSEN, A.; GRIMME, S. The Fractional Occupation Number Weighted Density as a Versatile Analysis Tool for Molecules with a Complicated Electronic Structure. Chemistry - A European Journal, v. 23, n. 25, 2017.
DE CARO, D. et al. Four molecular superconductors isolated as nanoparticles. European Journal of Inorganic Chemistry, v. 2014, n. 24, 2014.
FERREIRA, G. B. et al. An experimental and theoretical study of the electronic spectra of tetraethylammonium [bis(1,3-dithiole-2-thione-4,5-dithiolato)zincate(II)], [NEt4]2[Zn(dmit)2], and tetraethylammonium [bis(1,3-dithiole-2-one-4,5-dithiolato)zincate(II)], [NEt4] 2[Zn(dmio)2]. Inorganica Chimica Acta, v. 359, n. 4, 2006.
FERREIRA, G. B. Espectroscopia Eletrônica de Complexos do 1,3-ditiola-2-tiona-4,5-ditiolato (dmit) e seu isólogo 1,3-ditiola-2-ona-4,5-ditiolato (dmio) com Elementos Representativos. Tese de Doutorado—Rio de Janeiro: Universidade Federal do Rio de Janeiro, 2007.
FERREIRA, G. B. et al. An experimental and theoretical study of the electronic spectra of tetraethylammonium [bis(1,3-dithiole-2-thione-4,5-dithiolato)M(III)] and tetraethylammonium [bis(1,3-dithiole-2-one-4,5-dithiolato)M(III)] (M = Sb or Bi). Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, v. 71, n. 1, 2008.
GIVAJA, G. et al. Electrical conductive coordination polymers. Chemical Society Reviews, v. 41, n. 1, 2012.
GRIMME, S. et al. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. Journal of Chemical Physics, v. 143, n. 5, 2015.
KATO, R. et al. Emergence of the Dirac electron system in a single-component molecular conductor under high pressure. Journal of the American Chemical Society, v. 139, n. 5, 2017.
KIRK, M. L.; MCNAUGHTON, R. L.; HELTON, M. E. The electronic structure and spectroscopy of metallo-dithiolene complexes. Progress in Inorganic Chemistry, v. 52, 2004.
NEESE, F. “Software update: the ORCA program system, version 4.0” . WIREs Computational Molecular Science, v. 8, n. 1, p. e-1834, 2018.
NEESE, F. Software update: The ORCA program system—Version 5.0 . WIREs Computational Molecular Science, n. November 2021, p. 1–15, 2022.
PAES, L. W. C. et al. Electronic structure and adsorption geometry of Pt and Pd metal complexes with 1,3-dithiole-2-thione-4,5-dithiolate ligand on TiO2(101) surface from first-principles calculations. Theoretical Chemistry Accounts, v. 138, n. 7, 2019.
PANTAZIS, D. A. et al. All-electron scalar relativistic basis sets for third-row transition metal atoms. Journal of Chemical Theory and Computation, v. 4, n. 6, 2008.
ROOS, B. O. et al. Multiconfigurational Quantum Chemistry. [s.l: s.n.].
VELHO, M. F. G.; SILVA, R. A. L.; BELO, D. The quest for single component molecular metals within neutral transition metal complexes. Journal of Materials Chemistry C, 2021.














