Autores
Peixoto, B.P. (UNIVERSIDADE FEDERAL FLUMINENSE) ; de Andrade, K.N. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Carneiro, J.W.M. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Fiorot, R.G. (UNIVERSIDADE FEDERAL FLUMINENSE)
Resumo
Compreender os fatores que controlam transformações químicas é essencial para
planejar e otimizar processos de interesse científico. Uma classe de reações
amplamente explorada nesse sentido é a substituição nucleofílica alifática,
etapas-chave em diversos processos com apelo industrial e biológico.Esta pode
ser descrita pelas vias SN1 e SN2, compreendidas como extremos de um mecanismo
contínuo. A preferência por um caminho depende de diversos fatores, em especial,
estrutura do substrato e o solvente. Assim, o presente trabalho visa
racionalizar em nível DFT o mecanismo preferencial para substituições em
substratos que não se enquadram exclusivamente nos modelos SN1 e SN2. Grupos
adjacentes ao carbono eletrofílico demonstraram ter papel importante na
localização do mecanismo neste contínuo.
Palavras chaves
DFT; Substituição Nucleofílica; Mecanismos de Fronteira
Introdução
Dentre as maiores ambições dos profissionais da área de química, e também de
seus maiores desafios, estão o planejamento e a otimização de reações químicas.
Para o sucesso dessa tarefa, deve-se compreender os fatores que controlam a
reatividade química — mais precisamente, o mecanismo pelo qual as reações se
desenvolvem e como determinados aspectos estruturais conduzem a reação
preferencialmente por uma via em detrimento de outras. As reações de
substituições nucleofílicas em carbono sp3 compõem uma importante classe de
reações, constantemente presentes em etapas da síntese de substâncias com
diversas finalidades, como compostos tecnológicos e bioativos. Por estas razões,
estas reações têm sido amplamente exploradas do ponto de vista mecanístico. São
propostos dois modelos mecanísticos: substituição nucleofílica unimolecular
(SN1), ou DN+AN (DN — dissociação do grupo de saída, AN — associação do
nucleófilo), e substituição nucleofílica bimolecular (SN2), ou ANDN. Ambos podem
ser admitidos como extremos de um mecanismo contínuo e entre eles são propostos
mecanismos de fronteira. (HAMLIN et al., 2018). Devido à diversidade estrutural
de substâncias químicas aptas a essas reações, os modelos muitas vezes não são
suficientes para justificar resultados cinéticos e a estrutura dos produtos
formados. A elucidação dos fatores que controlam os mecanismos de uma reação
química permite o ajuste racional e o planejamento rotas sintéticas mais
eficientes. (VERMEEREN et al., 2021) Neste sentido, o presente trabalho objetiva
compreender com maior profundidade como diferentes estruturas de substratos
influenciam os mecanismos desta importante classe de reações, por meio da
identificação de vias de reação de menor energia em função das características
estruturais e eletrônicas.
Material e métodos
Cálculos computacionais de otimização de geometria e frequências vibracionais
foram realizados no software Gaussian 09 em nível da Teoria do Funcional da
Densidade (DFT), com o método computacional M06-2X/au-cc-pVTZ, previamente
validado de acordo com parâmetros geométricos e energéticos.
Para determinar o mecanismo preferencial (SN1 ou SN2), avaliamos substratos
cujos carbonos eletrofílicos não se encaixam exclusivamente nos modelos
mecanísticos limites: secundário (A), secundário congestionado (B), alílico (C),
benzílico (D) e α-heteroátomos (E) – Figura 1a. Para avaliar exclusivamente
aspectos cinéticos, minimizando influências termodinâmicas na identificação do
mecanismo preferencial, foram simuladas reações identidade, tendo cloreto como
nucleófilo (ClNu) / grupo de saída (ClGS). O efeito do solvente também foi
examinado para alguns substratos empregando-se o modelo do contínuo polarizável
(PCM), ao considerar a formamida implicitamente como solvente. Por fim, foi
avaliada a natureza associativa/dissociativa dos estados de transição (TS)
utilizando o diagrama de More O’Ferrall Jencks das ordens de ligação C-ClGS e C-
ClN, calculadas com NBO (do inglês, Natural Bond Orbital).
Resultado e discussão
O mecanismo bimolecular mostrou-se preferível para a maioria dos substratos, com
barreiras consideravelmente menores em fase gás e em solvente (Figura 1b) em
relação à energia para formação do carbocátion. Apenas para 1Ei a via
unimolecular é preferida. Nesta, a estrutura referente ao estado de transição
SN2 foi otimizada como um intermediário, possivelmente devido à dispersão da
carga positiva do átomo de nitrogênio que confere ao carbocátion um caráter de
imínio. Os resultados obtidos simulando solvatação implícita evidenciam a
influência do solvente na estabilização de íons, havendo diminuição
significativa da diferença de energia entre o caminho SN1 e SN2. Comparados aos
substituintes alquila (1A e 1B), a inclusão de sistema π vizinho ao centro
eletrofílico (1C e 1D) promoveu redução da barreira energética em
aproximadamente 5 Kcal mol–1. Para os sistemas com α-heteroátomos (E), observou-
se que a substituição com –OMe apresentou a menor barreira de energia via SN2,
enquanto o –PHMe apresentou a maior.
O diagrama de More O’Ferrall Jencks (Figura 2) apresenta resultados consistentes
com os discutidos anteriormente. O ponto estacionário localizado para 1Ei
apresenta alto caráter dissociativo, confirmando a natureza de um intermediário
carbocátion/imínio, esperado para uma via SN1. No outro extremo, próximo ao
caminho concertado, encontra-se o estado de transição 1Ev, com o grupo nitro em
α, que desestabiliza o caráter de carbocátion do TS. Assim, nucleófilo e grupo
de saída estão fortemente associados ao substrato, posicionando o estado de
transição mais próximo da linha diagonal tracejada, ANDN. Análises estão sendo
realizadas para compreender o efeito do substituinte na reatividade a partir do
modelo de interação-distorção.

(a) Estrutura dos substratos avaliados; (b) Valores de energia de ativação (SN2) e formação do carbocátion (SN1).
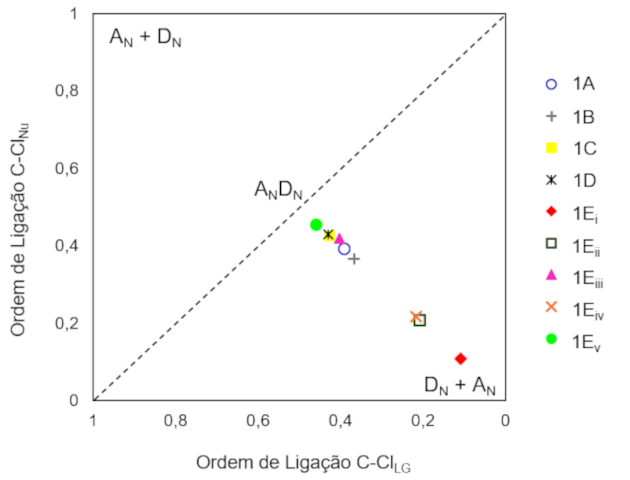
Diagrama de More O'Ferrall Jencks para pontos estacionários de mínimo (1Ei) e estados de transição (1A, 1B, 1C, 1D, 1Eii, 1Eiii, 1Eiv, 1Ev).
Conclusões
O caminho preferencial das reações de Substituição Nucleofílica Alifática foi
avaliado utilizando como modelo alguns substratos que não se encaixam
exclusivamente nos mecanismos uni- e bimolecular. As simulações revelaram que a
via SN2 é favorecida em relação a SN1 para a maioria dos substratos avaliados,
exceto para 1Ei. O efeito do solvente ficou evidente nessas reações, demonstrando
sua importância na estabilização dos íons presentes nas etapas reacionais. Por
fim, as análises do diagrama de More O’Ferrall Jencks corroboram as tendências das
barreiras calculadas.
Agradecimentos
CNPq, PROPPi-UFF
Referências
HAMLIN, T. A.; SWART M.; BICKELHAUPT, F. M. Nucleophilic Substitution (SN2): Dependence on Nucleophile, Leaving Group, Central Atom, Substituents, and Solvent. ChemPhysChem, 19, 1315– 1330, 2018.
VERMEEREN, P.; HAMLIN, T. A.; BICKELHAUPT, F. M. Chemical reactivity from an activation strain perspective. Chem. Commun., 57, 5880–5896, 2021














