Autores
La Torre, A.M.A. (IME) ; Almeida, J.S.F.D. (IME) ; Tanos, C.C.F. (IME)
Resumo
Este estudo teve como objetivo identificar potenciais inibidores da RNA
polimerase(RdRp) do SARS-CoV-2 através de estudos de modelagem molecular.Foram
realizados estudos de triagem virtual para obtenção de estruturas similares ao
composto de referência(remdesivir).Em seguida, as moléculas foram submetidas ao
programa PLANTS, com coordenadas 3D referentes ao sítio catalítico da proteína.
Foram realizados estudos de docking molecular no Molegro Virtual Docker 6.0 e os
resultados avaliados de acordo com a energia MolDock score e interação com
importantes resíduos catalíticos. Na sequência, realizou-se a predição dos
parâmetros toxicológicos com o auxílio do servidor PreADMET.Os resultados
servirão como ponto de partida para futuros estudos de dinâmica molecular e
experimentais.
Palavras chaves
SARS-CoV-2; COVID-19; Modelagem molecular
Introdução
Em dezembro de 2019, a Organização Mundial de Saúde (OMS) recebeu alertas sobre
um surto de pneumonia atípica em Wuhan, na China. A princípio medidas de
isolamento foram tomadas.Os pesquisadores denominaram a doença como COVID-19,
causada pelo coronavírus SARS-CoV-2 e considerada potencialmente grave, de
elevada transmissibilidade e com disseminação global(TILLOCA et al, 2020).
Em março de 2020, a Organização Mundial de Saúde (OMS) declarou a COVID-19 como
uma pandemia, que foi capaz de modificar as relações internacionais, familiares
e socioafetivas, além de mudanças profundas na economia dos países.
Em todo o mundo, mais de 6 milhões de pessoas já morreram em decorrência da
doença, e o número de casos confirmados ultrapassa 600 milhões de acometidos,
segundo dados obtidos a partir da OMS(2022).
De acordo com WANG(2020), os coronavírus(CoVs) empregam um mecanismo com
múltiplas subunidades para replicação e transcrição de seus genomas de RNA.
Portanto, a RNA polimerase dependente de RNA (RdRp) é considerada um alvo
promissor para inibidores antivirais, como o remdesivir, que catalisa a síntese
de RNA viral desempenhando um papel central no bloqueio do ciclo de replicação e
transcrição do coronavírus(GAO et al, 2020).
A partir dos avanços nas pesquisas, a flexibilização do uso de máscaras, o
desenvolvimento de vacinas contra o coronavírus e as campanhas de vacinação em
massa que estão em curso, comprovam, a cada dia, a redução efetiva da
mortalidade de pacientes infectados pelo vírus(RIVASE et al, 2021)
Apesar da vacina representar a melhor forma de proteção contra a COVID-19,torna-
se de suma importância destacar as limitações atreladas a elas, tais como
imunização de curto prazo, necessidade de doses de reforço, reações alérgicas
graves e efeitos colaterais de longo prazo, ainda desconhecidos(KURUP et
al,2021).
Além disso, deve-se considerar que a taxa de vacinação ainda é reduzida em
muitos países de baixa e média renda, ademais, ainda há grande parte da
população infantil sem acesso às vacinas contra COVID-19 em todo o mundo.
Assim como, pessoas sem acesso a qualquer vacina, ou seja, que podem contrair o
SARS-CoV-2 e desenvolver a forma mais grave da doença (MA et al, 2020).
A despeito do impacto epidemiológico, o tratamento da COVID-19 ainda não está
elucidado(WEN et al, 2022).
Segundo RAMOS et al(2022), na luta contra o coronavírus faz-se necessário
acelerar a descoberta de novos fármacos, por isso, o reposicionamento de
medicamentos já conhecidos representa uma boa estratégia.
O remdesivir é um pró-fármaco análogo de nucleotídeo de ação direta que
inibe o RNA ao incorporar trifosfatos e interferir na atividade da RNA
polimerase viral, foi aprovado pela agência americana Food and Drug
Administration(FDA) para a terapia contra COVID-19(GORDON, 2020).
Inúmeros estudos já comprovaram que o remdesivir é um potente antiviral contra
SARS-CoV-2(FERRARA et al, 2021).Portanto, torna-se indispensável destacar a
utilização de técnicas computacionais, que apresentam capacidade de explorar, in
silico, informações sobre alvos específicos que venham a favorecer o
reaproveitamento de fármacos, revolucionando o desenvolvimento de novas terapias
para combater a COVID-19 de maneira altamente específica e eficiente (ALMEIDA et
al, 2021).
Visando encontrar moléculas com potencial para inibir a replicação viral do
SARS-CoV-2, o remdesivir foi utilizado como molécula referência neste estudo de
modelagem computacional.
Foi realizada uma triagem virtual em uma biblioteca contendo medicamentos
aprovados pela Food and Drug Administration (FDA), em seguida, esses compostos
foram rastreados a partir do banco de dados PubChem, com a seleção das moléculas
com melhor pontuação para um ancoramento molecular utilizando o Molegro Virtual
Docker(MVD ®)6.0(THOMSEN & CHRISTENSEN, 2006).
As moléculas obtidas foram avaliadas de acordo com valores de energia (MolDock
score) e interação com importantes resíduos catalíticos. Na sequência, testes in
silico de previsão de toxicidade foram realizados usando o servidor online
PreADMET, disponível em (https://preadmet.bmdrc.kr/).
Os parâmetros de toxicidade incluíram carcinogenicidade em roedores,
mutagenicidade, teste Ames e inibição do gene HERG.
Devido à urgência de estratégias eficazes para o tratamento da COVID-19, o
reaproveitamento de medicamentos antivirais é relatado na literatura por
apresentar grandes vantagens, pois a farmacocinética, farmacodinâmica e perfis
de segurança desses medicamentos já estão bem estabelecidos(RAMOS et al, 2022).
Portanto, a aplicação de métodos in silico (triagem virtual, análise PreADMET,
bem como docking e dinâmica molecular são ferramentas valiosas para pesquisar o
espaço químico e selecionar as melhores estruturas que poderiam ser empregadas
em testes in vivo, minimizando o tempo e gastos com investimentos para encontrar
o tratamento da infecção por SARS-CoV-2(SILVA et al, 2022).
Material e métodos
Seleção de moléculas por ligand-based virtual screening(LBVS) e preparação dos
Ligantes
O banco de dados PubChem(KIM et al, 2019), com mais de 110 milhões de compostos
foi utilizado como recurso para a busca de moléculas similares ao remdesivir(GS-
5734), utilizado como molécula referência neste estudo.
Modelo da proteína
O modelo do receptor utilizado foi construído a partir da estrutura
cristalográfica da RdRp(código PDB: 7BV2,resolução de 2,50 Å).O arquivo PDB
(Protein Data Bank, https://www.rcsb.org/)foi submetido ao servidor
Swiss Model Server (referência) e em seguida foi validado utilizando-se o
servidor PDBsum(http://www.ebi.ac.uk/thornton-srv/databases/cgi-
bin/pdbsum/GetPage.pl?pdbcode=index.html), onde foram obtidos o gráfico de
Ramachandran e outros parâmetros considerados satisfatórios para a validação do
modelo (RAMACHANDRAN, 1968).
Ancoramento molecular
Detecção de cavidades da NSP12 e definição do espaço de busca (constrain)
As cavidades da NSP12 foram detectadas com MVD®6.0 (THOMSEN; CHRISTENSEN, 2006)a
partir da localização da forma trifosfato do remdesivir(RTP) cristalizado na
proteína e conhecimento do sítio ativo da NSP12, conforme descrito por Gao et al
(2020), pois o sítio ativo do domínio RdRp de SARSCoV-2 é formado por
subunidades estruturais A-G nos domínios palm e hand.
Onde a subunidade A é composta por resíduos 611-626(TPHLMGWDYPKCDRAM) e a
subunidade C contém os resíduos 753-767(FSMMILSDDAVVCFN).Em seguida, definiu-se
constrain onde foram geradas as poses a serem avaliadas pelo MVD®6.0. Este
espaço de busca foi definido de modo a consistir na menor esfera capaz de
englobar o sítio catalítico da NSP12.
Receptor-based virtual screening(RBVS) no servidor ChemoInfo
Para as etapas de simulação de ancoramento, a biblioteca de ligantes foi
submetida ao programa PLANTS (KORB; STÜTZLE; EXNER, 2009)onde o algoritmo se
baseia na otimização estocástica denominada “otimização tipo colônia de
formigas”, disponibilizado pelo servidor online ChemoInfo (DOUGUET, 2010,
2018).
Após esta etapa, os ligantes com pontuação igual ou superior a 80% da pontuação
máxima foram levados para posteriores estudos de ancoramento.
Ancoramento molecular com MVD 6.0
As moléculas aprovadas foram submetidas a trinta corridas, com o método “Virtual
Screening” selecionado para retornar apenas 20% das melhores poses. Cada pose
foi analisada quanto à energia de interação atribuída pelo algoritmo MolDock e
resíduos com as quais estabeleceu ligações de hidrogênio. As poses que não
estabeleceram ligações hidrogênio com pelo menos um resíduo do sítio catalítico
foram excluídas do trabalho. As poses restantes de cada ligante foram submetidas
a predição de toxicidade.
Predição dos parâmetros toxicológicos com o auxílio do servidor PreADMET
As moléculas que apresentaram desempenhos satisfatórios nas simulações de
ancoramento foram avaliadas quanto ao seu potencial toxicológico em relação a
carcinogenicidade em roedores, teste Ames e inibição hERG.
Resultado e discussão
Definiu-se um índice mínimo de similaridade de 80% em relação à molécula de
referência para a formação da biblioteca de ligantes. Essa similaridade foi
definida no banco de dados PubChem(KIM et al, 2019), utilizando-se o índice de
Tanimoto(VASS et al, 2016; WILDMAN, 2013).
O objetivo principal dessa triagem virtual foi identificar os compostos que
apresentam, no mínimo 80% de similaridade ao remdesivir e, portanto, maior
probabilidade de se ligar ao sítio ativo da NSP12 do SARS-CoV-2. As 1000
melhores moléculas ranqueadas após a triagem foram submetidas à RBVS, através do
servidor ChemoInfo (DOUGUET,2010, 2018) e pontuadas segundo o algoritmo CHEMPLP.
Para a realização do RBVS, o conhecimento prévio do sítio ativo da proteína, com
código PDB 7BV2, que apresenta o remdesivir cristalizado, associado a utilização
do MVD® (THOMSEN; CHRISTENSEN, 2006) permitiu a localização das cavidades da
NSP12 (subunidade catalítica da proteína), assim como, determinou as coordenadas
e raio do espaço de busca (raio igual a 12 Å, e centro localizado em x= 89,75;
y= 88,99 e z= 104,99) que foram utilizados para o ancoramento molecular.
Foram selecionadas 100 moléculas que apresentaram poses com pontuação PLANTS
menor ou igual a -137,66 kcal/mol, o equivalente a no mínimo 80% da melhor
pontuação apresentada, além do remdesivir, que apresentou pontuação -89,39
kcal/mol. Sendo nomeadas lig1 a lig100, de acordo com a ordenação decrescente da
pontuação obtida no ChemoInfo e o remdesivir, para as etapas seguintes.
Das 100 moléculas submetidas a estudos de ancoramento no MVD® (THOMSEN;
CHRISTENSEN, 2006), 51 poses foram obtidas, das quais apenas 16 exibiram poses
que formaram ligações de hidrogênio com pelo menos um resíduo catalítico da
NSP12, importante para a replicação como mostra a tabela 1.
Na sequência, o remdesivir e mais 16 moléculas, cujo CID está demonstrado na
tabela1, foram submetidas a predição dos parâmetros toxicológicos com o auxílio
do servidor PreADMET (https://preadmet.bmdrc.kr/), onde foram avaliadas quanto
ao seu potencial de carcinogenicidade em roedores, mutagenicidade com o teste
Ames e potencial para inibição do gene hERG, cuja avaliação pode ser conferida
na tabela 1.
Dentre as moléculas avaliadas, apenas quatro (lig21, lig30, lig51 e lig53)
apresentaram predição negativa em todos os parâmetros avaliados. Portanto, as
moléculas com algum potencial carninogênico ou mutagênico foi excluído do
trabalho.
Ainda sobre os parâmetros toxicológicos, avaliou-se também a possível capacidade
de inibição do gene hERG que está intimamente ligado com a função cardíaca. Este
parâmetro viabiliza a avaliação da influência do fármaco em teste na função
cardíaca, devido à capacidade do hERG de codificar a subunidade α do canal de
potássio presente no miócito.
Porém, a classificação mencionada (ambígua) em todos os ligantes pesquisados não
é clara quanto ao risco potencial, não podendo sozinha excluir ou confirmar o
risco cardíaco, devendo ser complementada com outros testes.
Por tratar-se de um estudo não clínico, a análise in silico não assegura a total
segurança toxicinética de um fármaco, fazendo-se necessária a utilização de
outros estudos da substância teste para maior segurança e confiabilidade dos
estudos.
Nos dados referentes a predição in silico realizada no PreADMET, o que confere
maior confiabilidade aos resultados é a utilização de dados do National
Toxicology Program (NTP) e do Food and Drug Administration (FDA), através de
aplicação de algoritmos. Além disso, a aplicação do servidor possibilita a
redução de custos e tempo de análise.

CID e resultados quanto a predição de toxicidade, interação com resíduos importantes e energia MolDock de 16 ligantes e remdesivir
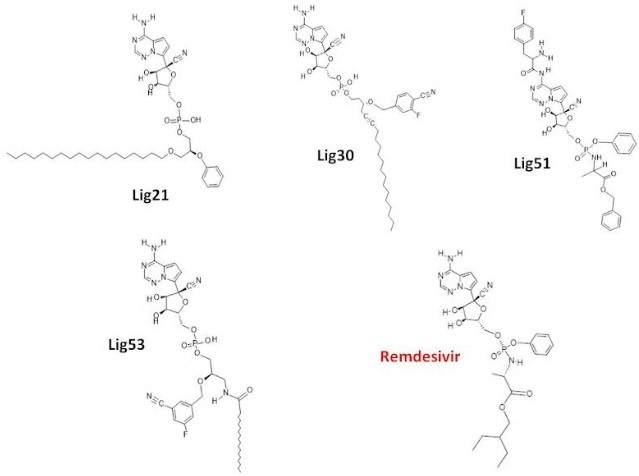
Estruturas dos quatro ligantes que apresentaram resultados satisfatórios e remdesivir
Conclusões
Desde o início da pandemia de COVID-19 houve uma enorme mobilização de
pesquisadores em todo o mundo promovendo pesquisas e ensaios clínicos na tentativa
de encontrar tratamentos para a infecção por coronavírus. O investimento na
pesquisa de reposicionamento de fármacos aumentou sensivelmente, e a utilização de
ferramentas computacionais como triagem in sílico mostrou-se bem-sucedida na
identificação de compostos ativos contra alvos moleculares importantes do SARS-
CoV-2. Os resultados obtidos até o presente momento sugerem que dentre uma
biblioteca de 1000 moléculas similares ao remdesivir, apenas 4 (quatro)
apresentaram resultados favoráveis. Os ligantes (21,30, 51 e 53) atenderam aos
critérios de seleção, e, ainda, mostraram-se satisfatórios nos estudos de
toxicidade. Estes ligantes serão submetidos em uma etapa subsequente a estudos de
dinâmica molecular e cálculos de MM-PBSA para corroboração dos resultados de
ancoramento já obtidos.
Agradecimentos
Agradeço ao Instituto Militar de Engenharia e ao Laboratório de Modelagem
Molecular aplicada à Defesa Química e Biológica do IME por fornecer todos os
recursos necessários para a realização deste trabalho.
Referências
ALMEIDA, Joyce SFD et al. Searching for potential drugs against SARS-CoV-2 through virtual screening on several molecular targets. Journal of Biomolecular Structure and Dynamics, v. 40, n. 11, p. 5229-5242, 2022.
DOUGUET, Dominique. Data sets representative of the structures and experimental properties of FDA-approved drugs. ACS medicinal chemistry letters, v. 9, n. 3, p. 204-209, 2018.
DOUGUET, Dominique. e-LEA3D: a computational-aided drug design web server. Nucleic acids research, v. 38, n. suppl_2, p. W615-W621, 2010.
FERRARA, Francesco et al. Remdesivir and COVID-19. Irish Journal of Medical Science (1971-), v. 190, n. 3, p. 1237-1238, 2021.
GAO, Yan et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science, v. 368, n. 6492, p. 779-782, 2020.
GORDON, Calvin J. et al. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. Journal of Biological Chemistry, v. 295, n. 20, p. 6785-6797, 2020.
KIM, Sunghwan et al. PubChem 2019 update: improved access to chemical data. Nucleic acids research, v. 47, n. D1, p. D1102-D1109, 2019.
KORB, Oliver; STUTZLE, Thomas; EXNER, Thomas E. Empirical scoring functions for advanced protein− ligand docking with PLANTS. Journal of chemical information and modeling, v. 49, n. 1, p. 84-96, 2009.
KURUP, Drishya; SCHNELL, Matthias J. SARS-CoV-2 vaccines—the biggest medical research project of the 21st century. Current Opinion in Virology, v. 49, p. 52-57, 2021.
MA, Cuiqing et al. From SARS-CoV to SARS-CoV-2: safety and broad-spectrum are important for coronavirus vaccine development. Microbes and Infection, v. 22, n. 6-7, p. 245-253, 2020.
RAMACHANDRAN, G. N.; SASISKEHARAN, V. Conformation of polypeptides and proteins. Advances in Protein Chemistry, v. 23, p. 283-256, 1968.
RAMOS, Ryan S., et al. Identification of Potential Antiviral Inhibitors from Hydroxychloroquine and 1, 2, 4, 5-Tetraoxanes Analogues and Investigation of the Mechanism of Action in SARS-CoV-2. International Journal of Molecular Sciences, 2022, 23.3: 1781.
RIVASI, Giulia et al. Course and lethality of SARS-CoV-2 epidemic in nursing homes after vaccination in Florence, Italy. Vaccines, v. 9, n. 10, p. 1174, 2021.
SILVA, Rai C. et al. Natural products-based drug design against SARS-CoV-2 Mpro 3CLpro. International journal of molecular sciences, v. 22, n. 21, p. 11739, 2021.
THOMSEN, René; CHRISTENSEN, Mikael H. MolDock: a new technique for high-accuracy molecular docking. Journal of medicinal chemistry, v. 49, n. 11, p. 3315-3321, 2006.
TILOCCA, Bruno et al. Comparative computational analysis of SARS-CoV-2 nucleocapsid protein epitopes in taxonomically related coronaviruses. Microbes and infection, v. 22, n. 4-5, p. 188-194, 2020.
VASS, Márton et al. Molecular interaction fingerprint approaches for GPCR drug discovery. Current opinion in pharmacology, v. 30, p. 59-68, 2016.
WANG, Quan et al. Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell, v. 182, n. 2, p. 417-428. e13, 2020.
WEN, Wen et al. Efficacy and safety of three new oral antiviral treatment (molnupiravir, fluvoxamine and Paxlovid) for COVID-19: a meta-analysis. Annals of medicine, v. 54, n. 1, p. 516-523, 2022.
WILDMAN, Scott A. Approaches to virtual screening and screening library selection. Current pharmaceutical design, v. 19, n. 26, p. 4787-4796, 2013.
WORLD HEALTH ORGANIZATION (OMS). Coronavirus disease (COVID-19) pandemic. Disponível em:https://www.who.int/emergencies/diseases/novelcoronavirus-2019?clid=Cj0KCQjwtZH7BRDzARIsAGjbK2aaMW4rvDfjyDejjccymkKaR1gghGz2z6tFqK6IVhy53CjI1JYeExIaAsFUEALwwcB>. Acesso: 5 de setembro de 2022.














