Autores
Pacheco Arce, T.M. (UNIVERSIDAD ICESI) ; Álvarez Caballero, J.M. (UNIVERSIDAD DEL MAGDALENA) ; Coy Barrera, E. (UNIVERSIDAD MILITAR NUEVA GRANADA) ; Escorcia Cabrera, A.M. (UNIVERSIDAD ICESI)
Resumo
α-glucosidase inhibitors currently available for treatment of Type II diabetes
have undesired secondary effects. Therefore, the discovery of α-glucosidase
inhibitors with diminished side effects is of great importance. In this work,
molecular docking simulations were performed to model the formation of protein-
ligand complexes between α-glucosidase and dicoumaroylated flavonol rhamnosides
with experimentally proven inhibitory activity. The predicted complexes show
binding affinity values that correlate qualitatively well with the IC50
values determined experimentally. Structural analysis of these complexes led to the
identification of protein-ligand interactions that are key for the inhibitory
activity of the flavonol molecules.
Palavras chaves
Molecular docking; α-glucosidase; Diabetes
Introdução
There are about 425 million people with diabetes worldwide, Type II diabetes being
the most predominant. α-glucosidase inhibitors slow the release of free glucose
in the small intestines by inhibiting the hydrolysis of dietary carbohydrates, and
so are used for treatment of Type II diabetes. However, comercially available α-
glucosidase inhibitors such as acarbose induce undesired gastrointestinal side
effects, such as abdominal bloating, cramping, increased flatulence, or diarrhea.
Therefore, new α-glucosidase inhibitory drugs are required (Li et al, 2019;
Turner et al, 2020). Recently, a set of dicoumaroylated flavonol rhamnosides have been reported to
exhibit α-glucosidase inhibitory activities which are remarkably highger than
that exhibited by acarbose (Li et al, 2019). However, the action mechanism of
these molecules remains unknown. Therefore, in this study, we performed molecular
docking simulations to get molecular information on the binding mechanism of these
molecules and to identify the structural features that make them better inhibitors
than acarbose. The results of our simulations are expected to help to the rational
desing of new potent and selective α-glucosidase inhibitors with reduced side
effects.
Material e métodos
The compounds addressed in this study are shown in Figure 1. The structures
of these molecules were built with Marvin JS (Cherinka et al, 2019) and further optimized at the AM1 level
using the Gaussian09 program (Frisch et al, 2016). After optimization, the structures were submitted to the
DockThor server (Guedes et al, 2021) for docking simulations, with the crystal structure of α-glucosidase
with PDB ID 3TOP (Ren et al, 2011) as a target. The docking simulations were performed under default conditions
of the DockThor server. Ionizable protein residues were protonated considering standard protonation states
at pH 7. Several docking experiments using different docking boxes were performed for each
molecule. Except for compound 1, the α-glucosidase inhibitory activities (IC50) of
the compounds studied here have been reported previously (Li et al, 2019). Moreover, the 3TOP crystal
structure corresponds to the acarbose αglucosidase complex (Ren et al, 2011). Thus, acarbose was included in
this study as a reference system to validate our docking approach, while compound 1 is the first
attempt to use this approach as a predicting tool of inhibitory activity.
Resultado e discussão
The binding mode of acarbose in the 3TOP crystal structure and that predicted by
docking with the best score are alike (Figure 2). Moreover, the docking simulations show
that the flavonol compounds are able to form a higher number of complexes (9-14 complexes)
with α-glucosidase than acarbose (4 complexes). Furthermore, overall the complexes of the
flavonol compounds show higher binding affinity values (7.9 to -10.6 kcal/mol) than the
acarbose complexes (-7.1 to -9.0 kcal/mol). These results correlate qualitatively well with
the IC50 values determined experimentally. While the IC50 for
acarbose is 266.1 μM, the flavonol compounds 2-4 display IC50 values between
12 μM and 36 μM (Li et al, 2019). This shows the already known good performance of DockThor to
predict the structure of protein-ligand complexes and suggests that it can be used to
qualitatively assess/predict the αglucosidase inhibitory activity of compounds which have not
been experimentally tested. Thus, compound 1 is expected to exhibit an inhibitory
activity similar to compounds 2-4. Structural analysis of the complexes identified led to the identification of key protein-ligand interactions for α-glucosidase inhibitory activity.
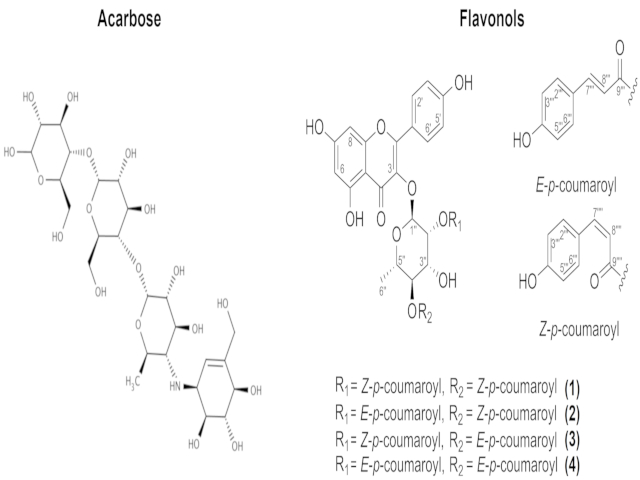
Compounds studied in this work.
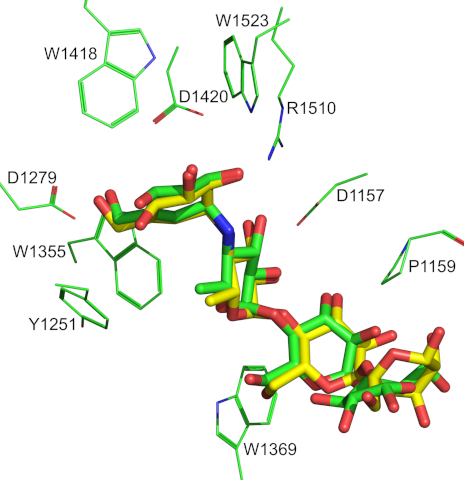
Superimposion of the α-glucosidase acarbose complex of the 3TOP crystal structure with the top scoring complex predicted by docking.
Conclusões
Dicoumaroylated flavonol rhamnosides are able to form a higher number of stable
complexes with α‑glucosidase and display higher binding affinities than acarbose, which explains the difference observed for these compounds regarding IC50 values. Our docking simulations provide information that may help to the rational design of more potent and selective α-glucosidase inhibitors.
Agradecimentos
Referências
CHERINKA, B.; ANDREWS, B. H.; SÁNCHEZ-GALLEGO, J.; BROWNSTEIN, J.; ARGUDO-FERNÁNDEZ, M.; BLANTON, M.; BUNDY, K.; JONES, A.; MASTERS, K.; LAW, D. R.; ROWLANDS, K.; WEIJMANS, A.; WESTFALL, K.; YAN, R. Marvin: A Tool Kit for Streamlined Access and Visualization of the SDSS-IV MaNGA Data Set. The Astronomical Journal, n. 2, 74, 2019.
FRISCH, M. J.; TRUCKS, G.; SCHLEGEL, H.; SCUSERIA, G.; ROBB, M.; CHEESEMAN, J.; SCALMANI, G.; BARONE, V.; MENNUCCI, B.; PETERSSON, G. Gaussian 09 D. 01, Wallingford CT 2016.
GUEDES, I. A.; BARRETO, A. M. S.; MARINHO, D.; KREMPSER, E.; KUENEMANN, M. A.; SPERANDIO, O.; DARDENNE, L. E.; MITEVA, M. A. New machine learning and physics-based scoring functions for drug discovery. Scientific Reports, n. 1, 3198, 2021.
LI, T.; KONGSTAD, K. T.; STAERK, D. Identification of α-Glucosidase Inhibitors in Machilus litseifolia by Combined Use of High-Resolution α-Glucosidase Inhibition Profiling and HPLC-PDA-HRMS-SPE-NMR. Journal of Natural Products, n. 2, 249-258, 2019.
REN, L.; QIN, X.; CAO, X.; WANG, L.; BAI, F.; BAI, G.; SHEN, Y. Structural insight into substrate specificity of human intestinal maltase-glucoamylase. Protein & Cell, n. 10, 827-836, 2011.
TURNER, J.; THOMAS, L.; KENNEDY, S. Structural Analysis of a New Saccharomyces cerevisiae α-glucosidase Homology Model and Identification of Potential Inhibitor Enzyme Docking Sites. Journal of Young Investigators, n. 4, 27-33, 2020.














