Autores
Magalhães, L.F. (UFSJ) ; Cunha, L.R.C. (UFSJ) ; Padilha, L.A. (UNICAMP) ; Vale, B.R.C. (UNICAMP) ; Schiavon, M.A. (UFSJ)
Resumo
Os nanocristais (NCs) de perovskitas apresentam curto tempo de vida radiativo,
limitando seu potencial em aplicações de transferência de carga e de energia.
Para obter um estado excitado de vida longa, a transferência de energia tripleto
(TET) para moléculas orgânicas foi desenvolvida para armazenar a energia de
excitação de NCs em tripletos moleculares de vida longa. Sintetizou-se NCs
cúbicos e preparou-se uma solução de R6G, contendo dímeros. A fotoluminescência
resolvida no tempo da mistura indicou um quenching dinâmico que ocorre por FRET.
Portanto, após a excitação, os NCs [CsPbBr][/3] transferem sua energia via FRET
para o estado excitado singleto dos dímeros R6G, relaxando rapidamente para o
estado tripleto, o qual foi confirmado utilizando-se espectroscopia de absorção
transiente
Palavras chaves
Perovskita; FRET; Transferência de Energia
Introdução
As perovskitas constituem uma classe de compostos de fórmula geral [ABX][/3], em
que A e B são cátions metálicos de diferentes tamanhos e X é um ânion,
geralmente um óxido [O][/2-] ou íons haletos ([Cl][/-], [Br][/-], [I][/-]) (LIU
et al, p. 9302, 2019). São inúmeras as possibilidades de formação de estruturas
do tipo perovskita, sendo que algumas alterações nessas formações trouxeram
grandes avanços, como a substituição de cátions orgânicos por metais, como o
césio, aprimorando a estabilidade do material que passou a resistir a umidade e
a decomposição sob altas temperaturas. (SHOAIB et al, p. 15594, 2017)
Dentre essa classe, os nanocristais de perovskitas de haleto de chumbo (NCs)
surgiram como materiais atraentes na captação e emissão de luz para muitas
aplicações, caracterizados por apresentarem amplo espectro de absorção com alto
coeficiente de absorção ([1O][/5] [mol][/-1]L [cm][/-1]). No entanto, o tempo de
vida radiativo desses NCs é de poucos nanossegundos à temperatura ambiente, o
que pode limitá-los a aplicações relacionadas a carga e transferência de
energia. Para obter um alto coeficiente de absorção e um estado excitado de
longa duração, a transferência de energia tripleto (TET) para moléculas
orgânicas foi desenvolvida como um meio eficaz de armazenar energia de excitação
de NCs em tripletos moleculares de longa duração. (HE et al, p.2786, 2021)
Uma das aplicações da sensibilização de tripletos é no processo de conversão
ascendente de fótons de aniquilação tripleto-tripleto (TTA-UC), em que dois
estados tripletos em moléculas aniquiladoras vizinhas produzem, via acoplamento
eletrônico, um estado singleto de maior emissão. (LUO et al, p., 2020)
Recentemente, têm-se estudado TTA-UC usando NCs como doadores e moléculas
transmissoras com base no ácido 9-antraceno carboxílico. Embora muitas
abordagens tenham sido exploradas para aumentar o UCQY, os registros sempre
ficam entre 10 e 20%, mesmo que as TET de NCs para os transmissores sejam acima
de 80%. Conforme sugerido por Lai, R. et al, essa grande lacuna entre TET e UCQY
sugere limitações com os sistemas TTA-UC sensibilizados por NC. (LAI et al,
2020)
Dentre as diferentes abordagens para a sensibilização, a via de mecanismo Dexter
tem sido bastante empregada. Esta via requer uma grande sobreposição da função
de onda orbital entre o NC e a molécula aceptora, sendo eficiente somente para
distâncias muito curtas (<1 nm). Assim, para maximizar a eficiência de TET, os
transmissores são ancorados à superfície dos NCs. Contudo, esta abordagem
apresenta algumas desvantagens, como a necessidade de forte regime de
confinamento quântico para uma TET eficiente (LUO et al, 2019).
Para ampliar o UCQY, seria necessária uma grande distância de separação entre o
NC e o transmissor, tornando-se desafiadora a ocorrência via mecanismo de
Dexter. Por outro lado, a transferência de energia de ressonância de Förster
(FRET) é um processo que permite transferências em maiores distâncias (2 a 10
nm), mas requer grande sobreposição espectral da absorção e emissão de
receptores e doadores, respectivamente. (HOFMANN et al, p. 3, 2019).
FRET é um fenômeno eletrodinâmico, fundamentado na transferência de energia não
radiativa, decorrente de interações dipolo-dipolo de longo alcance entre o par
doador-aceptor, sendo necessária uma condição ressonante entre as oscilações dos
campos elétricos do estado excitado do doador e do estado fundamental do
receptor. De forma geral, o conceito por trás do FRET é a ideia de que a espécie
doadora sofre uma fotoexcitação externa, levando-a ao seu estado excitado,
induzindo um dipolo oscilante, gerando um momento de dipolo de transição na
molécula (µD). Assim, a espécie aceptora na vizinhança pode sentir o campo
elétrico oscilante, o que pode induzi-la também, a um momento de dipolo (µA),
devido à condição de ressonância. A transferência de energia acontece, de fato,
quando o doador retorna para seu estado fundamental, não oscilante, e o aceptor
começa a oscilar, apresentando µA, o que leva a molécula ao estado excitado.
Durante o processo, a espécie doadora excitada não emite um fóton discreto. Têm-
se um campo elétrico oscilante que apresenta um fóton virtual, havendo apenas a
interação elétrica ou coulômbica entre o par doador-aceptor. (VALEUR, 2001)
Para a sensibilização de tripleto, FRET é ineficiente pois a absorção de
tripletos é proibida por dipolo. Com base nisso, neste trabalho é proposto um
modelo que pode ser otimizado para TTA-UC usando o mecanismo de FRET. Foram
utilizados NCs [CsPbBr][/3] como doadores e dímeros de rodamina 6G (R6G) como
transmissores. Diferente da Rodamina B (RhB), a R6G não possui o grupo
carboxila, responsável por facilitar a ancoragem na superfície dos NCs como
demonstrado por Dubose et al (DuBose et al, 2021). Assim, a R6G foi crucial para
favorecer o mecanismo de FRET, além de seus dímeros do tipo H apresentarem uma
banda de absorção com forte sobreposição à PL de [CsPbBr][/3]
Material e métodos
Em um balão de três bocas, foram adicionados 0,0814 g [Cs][/2][CO][/3], 4 mL de
ODE e 0,25 mL de AO e a mistura foi seca sob vácuo durante 1 hora a 120 ºC. Em
seguida, injetou-se argônio ao balão a 150 ºC até completa reação do [Cs][/2]
[CO][/3] com AO. Para preparar as CsPbBr3 PQs, adicionou-se 0,069 g de [PbBr]
[/2], 5 mL de ODE, 0,5 mL de AO e 0,5 mL de OAm em um balão de três bocas, e a
mistura foi seca sob vácuo a 120 ºC por 1 hora. Passado 1 hora, a solução foi
aquecida até 150 ºC sob uma atmosfera de Ar. Logo após, injetou-se, rapidamente,
0,4 mL do oleato-Cs à 150 ºC, deixou reagir por 10 s e resfriou-se a solução em
banho de gelo. Após a etapa de síntese, transferiu-se a suspensão para um tubo
Falcon e adicionou-se 7,5 mL de álcool isopropílico, levando-o para a centrífuga
por 30 minutos a 9000 rpm. O sobrenadante foi retirado e as nanopartículas foram
redispersas em tolueno/hexano. Os nanocristais obtidos foram caracterizados por:
Espectroscopia UV/Vis, Espectroscopia de Fotoluminescência no estado
estacionário e resolvida no tempo, Difratometria de Raios X (DRX), Rendimento
quântico de Fotoluminescência, Microscopia Eletrônica de Transmissão (TEM),
Espectroscopia de Absorção Transiente (TAS). Também preparou-se uma solução
concentrada de Rodamina 6G em etanol (0,02 mol [L][/-1]). Em seguida, realizou-
se duas diluições desta em tolueno, sendo a primeira à uma concentração de 0,012
mol [L][/s-1] e a segunda conforme a concentração desejada para cada
experimento, de forma a obter uma solução final que continha a maior quantidade
possível de rodamina em tolueno e a menor quantidade de etanol.
Para o
experimento de transferência de energia de ressonância titulou-se uma suspensão
de [CsPbBr][/3](10, 20, 40, 60, 80,100, 120 e 140 μL) à uma alíquota de 2500 μL
da solução de rodamina 6G na concentração 1,5 μmol [L][/-1]. Os espectros de
emissão foram registrados sob excitação à 400 nm. Para a construção da curva de
Stern-Volmer, preparou-se soluções com diferentes concentrações de R6G (0,5;
1,0; 1,5; 2,0; 2,5; 3,0; 3,5; 5,0; 7,0 e 10,0 µmol [L][/-1]) em tolueno sob uma
concentração fixa de perovskita. Foram realizadas, medidas de fotoluminescência
e de absorção UV destas amostras. Plotou-se a razão da intensidade de
fluorescência no estado estacionário na ausência e presença do aceptor (I/I0 )
em função da concentração deste último.
Realizou-se também experimentos de ancoragem e para isso, o complexo [CsPbBr]
[/3]-R6G foi preparado pela adição de 0,015 g de pó de R6G em 5 mL da solução do
NC em hexano, sendo sonicado por 30 minutos. A mistura foi filtrada em membrana
de PVDF CHROMAFIL com poros de 0,20 µm. Esta solução foi analisada por meio de
espectroscopia UV-Vis e de fotoluminescência no estado estacionário e resolvida
no tempo, utilizando uma cubeta de quartzo (Hellma) de 2 mm de caminho óptico.
Para fins comparativos, realizou-se o mesmo experimento para formação do
complexo [CsPbBr][/3]-R6G.
Resultado e discussão
Os espectros dos NCs de perovskitas podem ser visualizados na Figura 1a, os
quais apresentaram bandas de absorção e emissão em 493 e 507 nm,
respectivamente. O rendimento quântico de fotoluminescência foi determinado a
partir de um padrão de Fluoresceína (ϕf=92%), encontrando-se 55,5 %. Por medidas
de difração de Raios X, identificou-se três picos principais, relativos ao
sistema cristalino de fase cúbica Pm-3m. Realizou-se medidas de TEM, Figura 1a,
as quais confirmaram a obtenção dos NCs de forma cúbica, com distribuição de
tamanho centrada em 8nm, a qual concorda bem com o máximo do espectro de
absorção. Como o raio de Bohr do éxciton de [CsPbBr][/3] é de 7 nm, esses NCs
estão no regime de confinamento quântico moderado-fraco.
Os espectros de absorção e emissão da Rodamina 6G dispersa em tolueno, 1,5 µmol
[L][/-1], Figura 1b, mostram uma banda principal de emissão em 559 nm e duas
bandas de absorção, uma em 540 nm e outra em 513 nm, o que denota a formação de
dímeros. Pela posição da banda do dímero, conclui-se que este é do tipo H.
Embora a presença do dímero seja clara no espectro de absorção, indicando que
sua formação ocorre no estado fundamental, sua contribuição no espectro de
emissão não é evidente. Para avaliar esta contribuição, registrou-se espectros
de emissão e excitação de R6G em tolueno em diferentes concentrações (1,8 a 48
µmol [L][/-1]), além de um espectro de PL resolvido no tempo. O espectro de
emissão apresenta uma única banda principal, mas é possível observar um ombro na
região de 620 nm que tende a intensificar com o aumento da concentração,
indicando que esta emissão tenha contribuição do dímero.
Na Figura 1c são apresentados os espectros de excitação, monitorando-se a
emissão em 580 nm. Por ele se observa claramente duas bandas, uma localizada em
539 nm e outra em 510 nm. Foram realizados, também, experimentos provando-se a
emissão em 620 nm, no entanto, o perfil do espectro de PLE é muito semelhante ao
medido em 580 nm. Este fato sugere que existe uma sobreposição espectral entre
as emissões do dímero e do monômero. Para os casos em que os PLQYs das duas
espécies são da mesma ordem, ao se provar a emissão em 620 nm, era esperado que
esta transição se sobressaísse em relação a outra. Como isto não foi observado,
acredita-se que o PLQY do monômero seja muito maior que o do dímero, mas que
existe uma concentração muito maior deste último em relação ao primeiro.
A Figura 1d contém espectros de fotoluminescência resolvido no tempo da R6G (40
µmol [L][/-1]), para diferentes tempos de atraso. Nota-se que para tempos de
atraso de 0 a 0.5 ns, existem duas bandas de emissão, uma em 560 nm e uma em 620
nm, sendo que esta última decai para tempos acima de 1 ns. Na Figura 1e são
apresentadas curvas de decaimento de fotoluminescência da R6G em diferentes
comprimentos de onda. As curvas monitoradas em 510 e 620 nm são idênticas entre
si e de natureza biexponencial. Isso porque, como o dímero e o monômero
contribuem na emissão um do outro, é razoável pensar que ambos contribuem também
no decaimento. Deste modo, realizou-se um ajuste da função biexponencial
conforme a equação 1: PL(t)= A1 exp(-t/τ1)+ A2 exp(-t/τ2).
Pelo ajuste obteve-se uma constante de tempo de 0,63 ns, a qual, em função da
evidência experimental de que a banda em 620 nm decai em menos de 1 ns, foi
atribuída ao dímero, e 2,9 ns, pertencente aos monômeros. Por ser menos
emissivo, esperava-se que o dímero tivesse um maior tempo de vida, mas não foi o
observado, sugerindo a presença de um processo não-radiativo muito eficiente que
compete com a taxa de decaimento radiativo. Assim avaliou-se os níveis de
energia dos monômeros e dímeros da R6G, conforme estudos de Chibisovi (CHIBISOV
et al, 1999). Os dímeros, após fotoexcitação, passam por conversão interna de
[S][/1,H] para [S][/1,J], resultando em uma redução energética significativa do
estado singleto e tripleto dos dímeros, quando comparados aos monômeros,
favorecendo uma taxa de cruzamento intersistema. Portanto, o curto tempo de vida
de fotoluminescência do dímero de R6G pode ser atribuído ao eficiente processo
de cruzamento intersistema.
Como o espectro de absorção do dímero se sobrepõe ao espectro de emissão dos NCs
de [CsPbBr][/3], Figura 1f, é possível que exista um processo de transferência
de energia quando ambos interagirem. Sabendo-se disso, calculou-se o raio
crítico de Förster (R0), obtendo-se o valor de 5,7 nm.
Na Figura 2a são apresentados espectros de emissão dos NCs na ausência e
presença de diferentes concentrações de R6G, excitados em 400 nm. Neste caso, à
medida que a concentração de R6G aumenta, a intensidade de fotoluminescência dos
NCs é extinguida e, juntamente, a intensidade de fotoluminescência da R6G é
ligeiramente aumentada. Este fato sugere que a transferência ocorre dos NCs para
R6G.
Para se obter informações da natureza do processo de transferência de energia
foi feito uma curva de Stern-Volmer. A Figura 2b mostra uma relação linear,
sugerindo que apenas um processo (estático ou dinâmico) está envolvido. Para se
confirmar qual a natureza da supressão, experimentos de fotoluminescência
resolvida no tempo, monitorados em 512 nm, foi empregada na presença e ausência
da R6G. Pela Figura 2c, nota-se que o decaimento da curva de fotoluminescência
da emissão dos NCs de [CsPbBr][/3] na presença de R6G é acelerado em relação ao
decaimento deles quando na ausência do aceptor. Com isso, conclui-se que o
processo de extinção é de natureza dinâmica, ou seja, o doador precisa estar no
estado excitado para que o aceptor receba a transferência de energia de forma
não radiativa.
Foi provado a fluorescência da R6G em 560 nm na ausência e presença dos NCs de
[CsPbBr][/3]. Na Figura 2d, a curva de decaimento da rodamina na presença dos
NCs decai mais rapidamente do que a curva para o decaimento da R6G. As curvas de
decaimento foram ajustadas com a função biexponencial, Eq. 1, revelando que, na
presença dos NCs, o componente relacionado ao dímero, torna-se majoritário, com
aumento da amplitude de 0,80 para 0,91 e do tempo de vida de 0,48 para 0,89 ns.
Este aumento no tempo de vida é uma evidência de FRET, sendo utilizado no
cálculo de eficiência do processo conforme a equação 2: (([τ][/Doador-Aceptor]/
[τ][/Aceptor]) -1)*100, obtendo-se 85%.
Foram conduzidos experimentos de espectroscopia de absorção transiente na escala
de tempo µs, com pulso laser de 355 nm para a excitação dos NCs, de forma a
sondar a presença do estado tripleto dos dímeros. Pela Figura 2e observa-se,
logo após a excitação do pulso laser, a depopulação do estado fundamental dos
NCs, com seu mínimo em 500 nm. Também se nota um sinal mais fraco em torno de
560 nm, atribuído ao estado singleto de R6G monômero/dímero, seguido por outro
sinal de magnitude semelhante em 660 nm, atribuído para o estado tripleto do
dímero R6G.
Foi realizado a tentativa de ancoragem de R6G na superfície dos NCs, para
avaliar a possibilidade de ocorrência do mecanismo de Dexter. Os espectros de
emissão dos NCs [CsPbBr][/3] puros e após o procedimento de ancoragem com R6G e
RhB, dispersos em hexano, são apresentados na Figura 2f. Nota-se que há,
basicamente, uma sobreposição das curvas de absorção da perovskita sem e após a
tentativa de ancoragem com R6G, diferentemente da ancoragem com RhB. Assim,
infere-se que o processo de ancoragem se deu somente pelas moléculas de RhB,
sugerindo a possibilidade de mecanismo de Dexter. Enquanto para R6G, os NCs de
perovskita encontram-se livres de R6G, visto que esta não apresenta grupamento
carboxila. Estes dados corroboram com a ideia de quenching por mecanismo de FRET
na interação [CsPbBr][/3]-R6G com sensibilização indireta do estado tripleto.
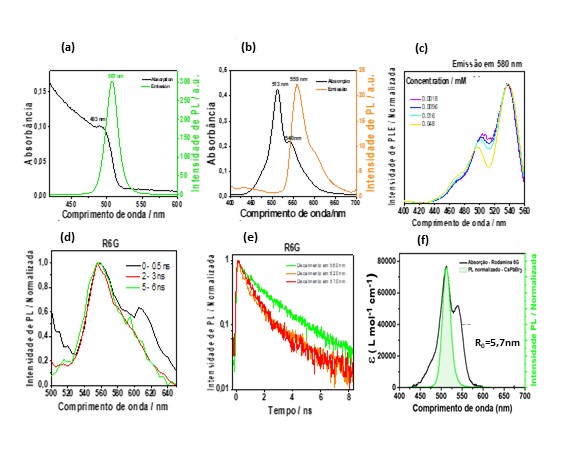
Espectro de PL e absorção(a) de CsPbBr3 e (b)R6G; Espectros de(c) excitação e (d) TR-PL e (e) Curvas de decaimento da R6G; (f) Sobreposição espectral
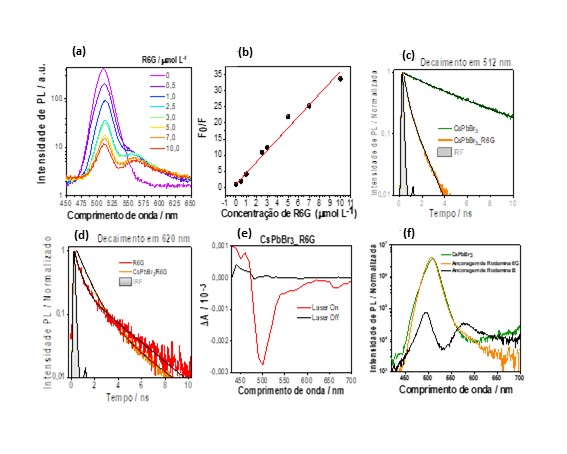
(a)PL CsPbBr3 com R6G(b)Curva de Stern- Volmer;Decaimento de PL de(c)CsPbBr3 e (d)R6G(e)TAS de CsPbBr3 /R6G(f)PL CsPbBr3 sob procedimento de ancoragem
Conclusões
Com este trabalho, foi possível a preparação e caracterização de perovskitas
inorgânicas de composição [CsPbBr][/3], bem como o estudo da interação entre esses
nanocristais e o corante Rodamina 6G. Concluiu-se que a formação dos dímeros na
solução de rodamina em tolueno foi de fundamental importância para que ocorresse
uma eficiente interação entre este corante e os NCs de perovskita. O mecanismo de
FRET, que foi caracterizado como um processo dinâmico, foi comprovado por meio de
estudos espectroscópicos, pelos quais pôde-se observar um aumento no tempo de vida
da espécie aceptora (dímero de R6G) de 0,48 ns quando na ausência do doador, para
0,89 ns quando em solução com os NCs, sendo este fator uma consequência do
processo de transferência de energia. Além disso, por meio de cálculos realizados,
obteve-se que R0 do dímero é de 5,7 nm e a eficiência de transferência foi de 85%.
Devido ao fato de o dímero apresentar uma proximidade energética entre os estados
S1 e T1, o que permite uma alta taxa do processo de cruzamento intersistema,
juntamente com o fato dessa espécie favorecer o processo de FRET, indiretamente, a
sensibilização do estado tripleto é favorecida, o que é relevante para aplicações
em fotocatálise.
Agradecimentos
Os autores agradecem ao CNPq, à CAPES e à FAPEMIG pelo apoio.
Referências
CHIBISOV, A. K.; ZAKHAROVA, G. V.; GÖRNER, H. Photoprocesses in dimers of thiacarbocyanines. Phys. Chem. Chem. Phys., v. 1, 1455-1460, 1999.
DUBOSE, J.T., KAMAT, P.V. Directing Energy Transfer in Halide Perovskite-Chromophore Hybrid Assemblies. Journal of the American Chemical Society, v. 143(45), 19214-19223, 2021.
HE, S., HAN, Y., GUO, J., WU, K. Long-Lived Delayed Emission from CsPbBr3 Perovskite Nanocrystals for Enhanced Photochemical Reactivity. ACS Energy Letters, v. 6 (8), 2786-2791, 2021.
Hofmann, Felix J. et al. Energy Transfer from Perovskite Nanocrystals to Dye Molecules Does Not Occur by FRET. Nano Letters, 1-7, 2019.
LAI, R., SANG, Y., ZHAO, Y., WU, K. Triplet Sensitization and Photon Upconversion Using InP-Based Quantum Dots. Journal of the American Chemical Society, v. 142 (47), 19825-19829, 2020.
LIU, Ronghuan et al. Low-reflection, (110)-orientation-preferred CsPbBr3 nanonet films for application in high-performance perovskite photodetectors. Nanoscale, v. 11 (19), 9302-9309, 2019.
LUO, X., LAI, R., LI, Y., LIANG, G., LIU, X., DING, T., WANG, J., WU, K. Triplet Energy Transfer from CsPbBr3 Nanocrystals Enable by Quantum Confinement. Journal of the American Chemical Society, 141, 4186-4190, 2019.
LUO, X., LIANG, G., HAN, Y., LI, Y., DING, T., HE, S., LIU, X., WU, K. Triplet Energy Transfer from Perovskite Nanocrystals Enable by Quantum Confinement. Journal of the American Chemical Society,142, 11270-11278, 2020.
SHOAIB, Muhammad et al. Directional Growyh of Ultralong CsPbBr3 Perovskite Nanowires for High-Erformance Photodetectors. Journal Of The American Chemical Society, v. 139 (44), 15592-15595, 2017.
VALEUR, B. Molecular Fluorescence: Principles and Applications, Wiley: VCH Verlag GmbH, 2001.














