Autores
Camargo-ayala, L. (UNIVERSIDAD DE TALCA) ; Bedoya, M. (UNIVERSIDAD CATÓLICA DEL MAULE) ; Prent-peñaloza, L. (UNIVERSIDAD ANDRÉS BELLO) ; Margarita, G. (UNIVERSIDAD DE TALCA) ; Gonzalez, W. (UNIVERSIDAD DE TALCA)
Resumo
The most frequent type of arrhythmia is atrial fibrillation (AF). The
electrophysiological behavior of the atrial is given by the action potential,
mediated by Nav1.5, Kv1.5, and TASK-1 channels, among other ion channels. A
strategy for the treatment of AF is the simultaneous inhibition of these three
channels using polipharmacological compounds. In this study, a rational compound
design was performed based on the scaffold structure of known local anesthetics.
After synthesis, the compounds were characterized, purified, and through
electrophysiology assays by the TEVC technique, the ability of the compounds to
inhibit these channels was tested. These new compounds capable of simultaneously
block Nav1.5, Kv1.5, and TASK-1 channels are a promising strategy for the
treatment of AF.
Palavras chaves
Atrial fibrillation; ion channels; polypharmaceuticals
Introdução
Atrial fibrillation (AF) is a prevalent type of arrhythmia and is a significant
cause of embolic stroke, heart failure, and cardiovascular morbidity. It has
been considered the most severe abnormal heart rhythm disorder. In normal
conditions, the electrophysiological behavior of the heart is given by a set of
ordered phenomena that result in rapid depolarization and slow repolarization,
which generates action potentials (AP). The AP of atrial cardiomyocytes is
initiated by membrane depolarization through Nav1.5 channels, a known
therapeutic target of AF. Potassium channels conduct repolarizing currents;
Kv1.5 channels, which are expressed in the atria but not in the ventricles, are
primarily responsible for the ultrafast potassium outflow current (IKur) and
determine the duration of atrial AP. Additionally, it has been suggested that
the K2P channels, TASK-1, may contribute to AP since they have been shown to be
inhibited by known Kv1.5 channel blockers, which has made them an unrecognized
target that could contribute to the clinical efficacy of these drugs against AF.
Consequently, the ion channels Nav1.5, Kv1.5, and TASK-1, constitute therapeutic
targets for AF. The simultaneous blockers of these channels could become
innovative strategies against AF, following the paradigm of polypharmacology.
The new drugs synthesized in this work are based on local anesthetics (LA). LA
exhibit antiarrhythmic capacity blocking atrial channels such as NaV1.5, Kv1.5
and TASK-1 channels, becoming potentially effective drugs for treating AF. The
primary purpose of this study was to obtain new N-aryl-2-(N-disubstituted)
acetamide-type molecules through organic synthesis by rational design based on
the common characteristics of LA compounds that block ion channels present in
the atrium.
Material e métodos
A common pharmacophore was obtained based on LA structures. These LA exhibit
antiarrhythmic properties and reported activity as atrial channels blockers. The
60 candidate compounds to be synthesized fulfilled the pharmacophoric
characteristics of LA. Once these candidates were proposed, in-silico studies
were carried out. Molecular coupling and binding energy calculations by the
Molecular Mechanics / Generalized Born Surface Area (MM-GBSA) method. The best
compounds were selected, considering three criteria; 1) the best binding free
energy, 2) interaction with residues reported as relevant for the binding of
local anesthetics, and 3) synthetic feasibility.
After selecting the 10 compounds with the best results in the in-silico
analysis, their synthesis was carried out, as follows; for the synthesis of
amides, N,N'-diisopropylcarbodiimide (DIC) was used as a coupling reagent,
followed by a nucleophilic substitution reaction. In general terms, (1 mmol, 1
equiv.) of 2-bromo acetic acid dissolved in DCM at 0°C was measured, and (1.1
mmol, 1.1 equiv.) of DIC was added at 0°C, subsequently reacted with different
aromatic amines (1.3 mmol, 1.3 equiv.), obtaining the intermediate amide product
which in turn was reacted with aliphatic amines (1.2 mmol, 1.2 equiv.), thus
obtaining the compounds in moderate to good yields, these compounds were
purified using column chromatography, and characterized by 1H-NMR, 13C-NMR, IR,
MS and HPLC. Then compounds were diluted in DMSO, and solutions were prepared at
a concentration of 100µM. Electrophysiological measurements were carried out
using the Two Electrode Voltage Clamp (TEVC) technique in Xenopus oocytes
systems. The inhibitory activity was evaluated of the 10 selected compounds
establishing their possible polypharmacological activity.
Resultado e discussão
A pharmacophore based on LA structures was proposed to establish the
characteristics that the compounds to be synthesized should have (Figure 1a).
The pharmacophoric characteristics of LA are 1) a hydrophobic domain, formed by
the alkyl substituents of the aromatic ring; 2) a hydrogen donor domain from the
amine nitrogen, 3) a hydrogen acceptor domain, the amide carbonyl oxygen, and 4)
a cation-forming domain, 60 initial compounds meeting these pharmacophoric
characteristics were designed. Through In-silico studios of molecular docking
and binding free energy calculations (MM-GBSA) of all the designed compounds
were performed, the 10 best compounds were selected and synthesized according to
the scheme in Figure 1b. Compounds with moderate to good yields (55-84%) (Figure
1c) were obtained. These compounds were purified and characterized using 1H-NMR,
13C-NMR, IR, and MS (Figure 2a). Once the identity and purity of the compounds
were fully corroborated, electrophysiology tests were carried out in Xenopus
oocytes using (TEVC) technique, to evaluate the possible polypharmacological
activity of the compounds. In general terms, most of the compounds block the
three channels Nav1.5, Kv1.5 and TASK-1. However, compounds 49, 14, and 18
showed the best activity (Figure 2b).
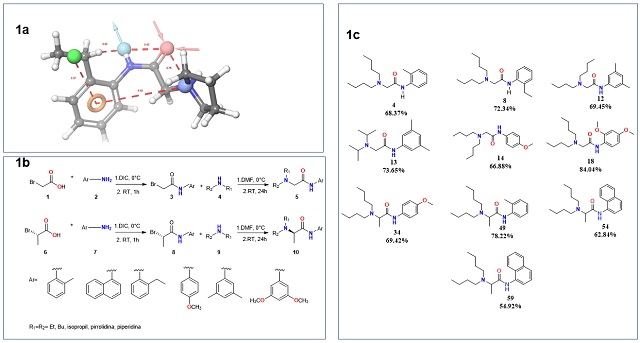
Rational design and synthesis of compounds. A. Proposed pharmacophore for LA. B. Scheme of synthesis. C. Synthesized compounds and yield.
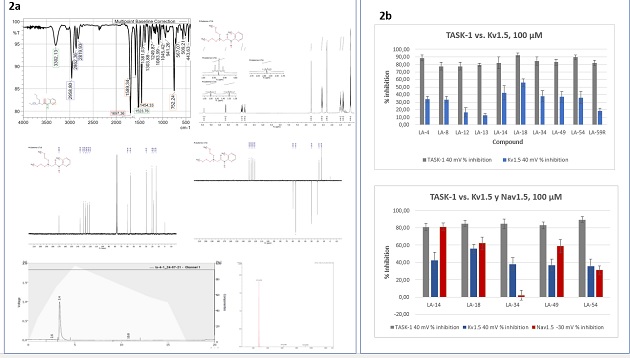
Characterization and biological tests of compounds. A. IR, NMR, MS and HPLC spectra. B. Inhibition plots at 100 µM.
Conclusões
Through rational design we proposed a series of 60 compounds based on the LA
pharmacophore with polypharmacological activity in atrial canals; Nav1.5, Kv1.5
and TASK-1. Of these, the 10 best were selected based on molecular docking
analysis and MMGBSA calculations. Once the synthesis of these compounds was
achieved, their ability to block their currents by TEVC was evaluated. Compounds
14, 18 and 49 had the best blocking activity in all three channels, this could be
a promising strategy for the treatment of AF.
Agradecimentos
Center for Bioinformatics, Simulation and Modelling (CBSM) and Laboratory of
Organic Synthesis and Biological Activity of the UTalca. Fondecyt Projects 1191133
and 1200531. ANID National Doctoral Scholarship 2019 Folio N°21190020.
Referências
(1) Ravens, U.; Poulet, C.; Wettwer, E.; Knaut, M. Atrial Selectivity of Antiarrhythmic Drugs.J. Physiol. 2013, 591 (17), 4087–4097. https://doi.org/10.1113/jphysiol.2013.256115.
(2) Kiper, A. K.; Rinné, S.; Rolfes, C.; Ramírez, D.; Seebohm, G.; Netter, M. F.; González, W.; Decher, N. Kv1.5 Blockers Preferentially Inhibit TASK-1 Channels: TASK-1 as a Target against Atrial Fibrillation and Obstructive Sleep Apnea? Pflugers Arch. Eur. J.Physiol. 2015, 467 (5), 1081–1090. https://doi.org/10.1007/s00424-014-1665-1.
(3) Nguyen, P. T.; DeMarco, K. R.; Vorobyov, I.; Clancy, C. E.; Yarov-Yarovoy, V. Structural Basis for Antiarrhythmic Drug Interactions with the Human Cardiac Sodium Channel. Proc. Natl. Acad.Sci. U.S.A. 2019, 116 (8), 2945–2954. https://doi.org/10.1073/pnas.1817446116.
(4) Chung, M. K.; Eckhardt, L. L.; Chen, L. Y.; Ahmed, H. M.; Gopinathannair, R.; Joglar, J. A.; Noseworthy, P. A.; Pack, Q. R.; Sanders, P.; Trulock, K. M. Lifestyle and Risk Factor Modification for Reduction of Atrial Fibrillation: A Scientific Statement From the American Heart Association. Circulation 2020, 141 (16), E750–E772. https://doi.org/10.1161/CIR.0000000000000748.














