MODELAGEM COMPUTACIONAL E ESTUDO SAR/QSAR DE DERIVADOS DE CHALCONA COM ATIVIDADE ANTICÂNCER DE PULMÃO
ISBN 978-85-85905-25-5
Área
Físico-Química
Autores
dos Santos Santos, A. (UEAP) ; Pinto Barbosa, J. (UEAP)
Resumo
As chalconas são moléculas orgânicas que têm obtido resultados significativos no tratamento de inúmeros tipos de câncer, por isso tem sido alvo de inúmeros estudos químicos e farmacológicos. A presente pesquisa viabilizou estudar computacional e quimiometricamente compostos derivados de chalconas contra o câncer de pulmão, e propor candidatos a fármacos, através de predição estatística e de ensaios in sílico (farmacocinética, toxicidade e docking molecular). Ao fim do presente estudo foi proposto 2 candidatos promissores a fármacos anticâncer de pulmão.
Palavras chaves
Câncer; Chalcona; In sílico
Introdução
O câncer é uma doença de proliferação celular em que células normais são transformadas por mutação genética em células com crescimento descontrolado. As células neoplásicas competem com as células normais pela obtenção de energia e nutrição, resultando em deterioração da função orgânica normal (GOLAN et al., 2012). De acordo com a Organização Mundial da Saúde (OMS), o câncer é atualmente responsável por uma em cada seis mortes no mundo. No Brasil, conforme o Instituto Nacional do Câncer (INCA) para 2016 estimou-se 17.330 casos novos de câncer de traquéia, brônquios e pulmões entre homens e 10.890 entre mulheres (INCA, 2016). No aspecto do tratamento do câncer, a quimioterapia é uma ferramenta clínica, que se caracteriza pela administração de drogas que irão combater os adenocarcionomas malignos instalados no corpo. Porém esse tipo de tratamento causa inúmeros efeitos colaterais e aumentam a resistência das células cancerosas, fazendo-se necessário buscar novos fármacos, que sejam mais eficazes no tratamento desse tipo de doença, e que não causem tantos efeitos adversos aos pacientes em tratamento. O objetivo central deste trabalho foi realizar o estudo computacional, que engloba a análise quimiométrica (QSAR), a aplicação da técnica de alinhamento e docking molecular, relacionando à estrutura-atividade (SAR), e propor modelos estatísticos capazes de fazer previsões de atividade de novas drogas anticâncer, bem como o estudo farmacológico dos compostos do treino e teste. Assim a presente pesquisa atribui enfoque as chalconas, moléculas pertencentes ao grupo dos flavonóides, que apresentam cadeia aberta, no qual dois anéis aromáticos são unidos por um sistema ceto-α,β-insaturado (ECHEVERRIA et al., 2009).
Material e métodos
A partir da literatura (Syam et al., 2012) investigou-se 20 compostos derivados de chalcona que compõem o conjunto treino. A hipótese estipulada foi que os compostos mais ativos apresentam IC50 ≤ 36,58 μg/mL. As estruturas foram desenhadas no programa Discovery Studio e otimizadas no programa Gaussian09 no método/base B3LYP/CEP31G**. Posteriormente foram calculados 1802 descritores moleculares através dos programas E-Dragon, Molekel, Marvin Sketch, PreADMET, Pharma Gist, Molispiration, e organizados em uma matriz de dados, a partir da qual foi realizado o estudo de QSAR (Relação Estrutura Atividade Quantitativa), onde foram utilizados os métodos estatísticos de PCA, HCA e KNN para a redução e qualificação dos descritores moleculares mais importantes. O estudo de SAR (Relação Estrutura Atividade) foi realizado no programa Pharma Gist e consistiu na busca de um modelo farmacofórico através da sobreposição das moléculas treino sobre a molécula mais ativa, para que alinhe o maior número de características em comum. Com o modelo farmacofórico, foi realizado a triagem virtual no programa ZINCPharmer com o intuito de buscar novos compostos com as características do farmacóforo base. A análise farmacocinética e toxicológica foi realizada no programa PreADMET com o conjunto treino e teste, para investigação dos processos de absorção, distribuição, metabolismo, eliminação e carcinogenicidade das drogas. O docking molecular foi realizado no programa Pyrx e consistiu em realizar a interação das drogas (conjunto treino e teste) ao receptor enzima quinase (código PDB: 2ITN) encontrada na literatura (Xian et al., 2014). Ao final foi realizada a predição estatística dos potenciais candidatos a fármacos anticâncer de pulmão, a partir do conjunto teste selecionado.
Resultado e discussão
A estatística multivariada PCA possibilitou separar o conjunto treino em mais ativos e menos ativos através dos descritores moleculares Mor11m, HOMO e ASA-. A técnica HCA e KNN confirmou esta separação. Através do alinhamento molecular do conjunto treino foi encontrada a região do farmacóforo que continha: 2 regiões aromáticas, 1 região aceptora de hidrogênio e outra hidrofóbica. Com auxílio desse modelo farmacofórico foi feito a triagem virtual e selecionou-se 10 moléculas derivadas de chalcona no banco de dados do programa ZINCPharmer e que compõem o conjunto teste. A análise farmacológica apresentou as propriedades farmacocinéticas e toxicológicas dos compostos estudados, e grande parte das moléculas apresentaram bom desempenho farmacocinético, e no aspecto toxicológico as moléculas menos ativas do conjunto treino foram as que em geral apresentaram caráter tóxico, bem como os compostos 21, 22 e 30 do conjunto teste. A aplicação da técnica de docking molecular proporcionou identificar a região de sítio ativo, em que poderiam ser acopladas as moléculas, assim todos os compostos obtiveram boas energias de afinidade com o sítio ativo da enzima quinase, a citar as moléculas mais ativas 4, 8 e 20 do conjunto treino e as moléculas 22, 24 e 30 do conjunto teste. Os orbitais de fronteira HOMO destacaram densidade na região da carbonila e dos anéis aromáticos, tanto para os compostos do conjunto treino quanto para os do teste, e essa característica é importante para a interação ligante receptor. Através dos modelos gerados pelo PCA e KNN, foi feita a predição quimiométrica, e dos 10 compostos do conjunto teste, apenas os compostos 24 e 26 (figura 1), foram apontados como mais ativos contra o câncer estudado.
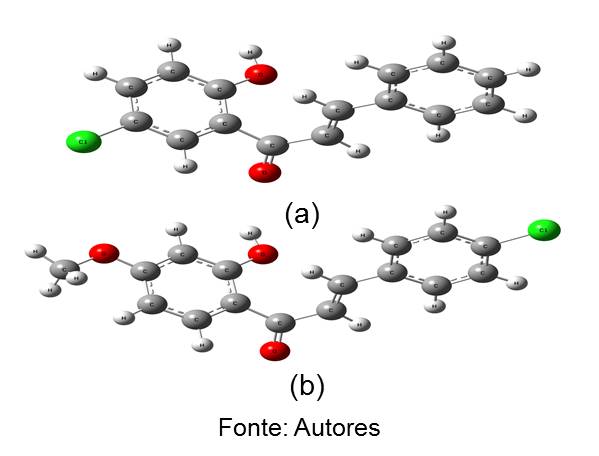
(a) comp. 24 (2E-1-(5-cloro-2-hidroxifenil)-3-fenilprop-2-en-1-ona); (b) comp. 26 (2E-3-(4-clorofenil)-1-(2-hidroxi-4-metoxifenil) prop-2-en-1-ona).
Conclusões
A técnica de PCA selecionou os descritores fundamentais para construção do modelo de predição de atividade. O estudo do orbital de fronteira HOMO permitiu identificar a região de interação da chalcona com o sítio ativo. O modelo farmacofórico auxiliou na triagem virtual para busca do conjunto teste. Os resultados da análise farmacológica foram importantes para avaliação da biodisponibilidade e toxicidade dos compostos, dessa forma tais resultados poderão ser usados como auxiliares no planejamento, síntese e ensaios biológicos de novos derivados de chalcona com atividade anticâncer de pulmão.
Agradecimentos
Ao CNPq, UEAP e CENAPAD-SP
Referências
INSTITUTO NACIONAL DO CÂNCER. Incidência de mortalidade de câncer no Brasil, 2016. Disponível em: http://www.inca.gov.br/estimativa/2016/index.asp?ID=2. Acesso 30/05/2017.
GOLAN, E. D.; TASHJIAN, A. H.; ARMSTRONG, E.J. Princípios da farmacologia: a base fisipatologica da farmacoterapia. 2ª ed. Rio de Janeiro: Guanabara Koogan, 2012.
SYAM, S.; ABDELWAHAB, S.I.; AL-MAMARY, M.; MOHAN, S. Synthesis of Chalcones with Anticancer Activities. J. Molecules. p. 6179-6195, 2012.
Xian-qiang, SUN; Lei, CHEN; Yao-zong, LI; Wei-hua, LI; Gui-xia, LIU; Yao-quan, TU; Yun, TANG. Structure-based ensemble-QSAR model: a novel approach to the study of the EGFR tyrosine kinase and its inhibitors. Acta Pharmacologica Sinica. 35: 301-310, 2014.
ECHEVERRIA, C., SANTIBANEZ, J.F., DONOSO-TAUDA, O., ESCOBAR, C.A. and Tagle, R.R. Structural antitumoral activity relationships of synthetic chalcones. Inter. Jour. Of Molec. Scien, 10, 221-23, 2009.








