QSAR e docagem molecular de derivados tetrahidronafto[1,2-b]azepinas utilizando a enzima dihidrofolato redutase de Trypanosoma cruzi
ISBN 978-85-85905-25-5
Área
Físico-Química
Autores
Cardoso, F.J.B. (UNIVERSIDADE FEDERAL DO PARÁ) ; Vilhena, K.S.S. (UNIVERSIDADE FEDERAL DO PARÁ)
Resumo
A relação quantitativa entre a estrutura de 21 derivados tetrahidronafto[1,2-b]azepinas e suas atividades foi avaliada pelo método dos mínimos quadrados parcias e o modelo QSAR proposto mostrou capacidade explicativa e predita aceitáveis, sendo que as atividades biológicas estão relacionadas com as propriedades atômicas (volume de van der Waals-Dv e eletronegatividade de Sanderson-HATS0e), eletrônica (GAP) e solubilidade (logP). A docagem molecular mostrou que os compostos interagem principalmente com o resíduo aromático Phe52 (interação π – π) e através de ligação de hidrogênio com Ser83 e Ile154 do sítio ativo da enzima dihidrofolato redutase de T. cruzi. O modelo matemático pode ser útil para a proposição de novos compostos anti-chagásicos com melhores afinidades pela enzima alvo.
Palavras chaves
QSAR; docagem molecular; dihidrofolato redutase
Introdução
A doença de Chagas é causada pelo protozoário parasito Trypanosoma cruzi sendo transmitida diretamente pelo vetor conhecido como barbeiro, ou também via transfusão sanguínea, transplante de órgãos, via congênita – de mãe para filho, ingestão de alimentos contaminados e exposição acidental em laboratório (CORTES-SERRA et al, p. 464, 2018). A infecção é considerada endêmica em países da América latina, com ocorrências registradas em outras localidades como Canadá, Estados Unidos, Espanha, Portugal, Japão e Austrália (WIGGERS et al, p. 2013). Devido ao crescente número de viagens e imigrantes, tem aumentado a preocupação com essa doença em áreas não endêmicas (CORTES-SERRA et al, p. 464, 2018). A enfermidade afeta cerca de oito milhões de pessoas ao redor do mundo, causando cerca de 14.000 mortes por ano. Além disso, provoca várias complicações médicas tais como dano ao músculo do coração (cardiomiopatia), sistema nervoso central e ao trato digestivo (megacólon e megaesôfago) consequentemente resultando em morte (SACCONNAY et al, p. e2689, 2014) Há apenas duas alternativas terapêuticas disponíveis para o tratamento da doença e são baseadas nos compostos nitroheterocíclicos, que são: o nifurtimox e o benzonidazol, sendo que ambos apresentam inúmeros efeitos colaterais, eficácia limitada e são efetivos somente na fase aguda da infecção (DINIZ et al, p. e00401, 2018). Dessa forma, o desenvolvimento de compostos mais efetivos para tratar a doença de Chagas é de grande importância. Uma linha de estudo que tem desempenhado papel fundamental no planejamento de novos compostos é o desenvolvimento de fármacos com a assistência de recursos computacionais, os quais podem ser classificados em métodos de planejamento de fármaco baseado na estrutura do ligante (Ligand-Based Drug Design - LBDD) e desenvolvimento de fármaco baseado na estrutura da macromolécula (Structure-Based Drug Design - SBDD). O método LBDD direciona sua análise em um conjunto de compostos com atividade biológica conhecida, obtendo características moleculares essenciais para a atividade, gerando regras e padrões representados por hipóteses farmacofóricas, modelos de classificação como os que relacionam quantitativamente estrutura-atividade (QSAR) e estrutura-propriedade (QSPR). Esses modelos são utilizados para predizer a atividade biológica, desenvolvimento de novos compostos e na triagem virtual baseada no ligante (Ligand-Based Virtual Screening - LBVS) (FERREIRA, OLIVA, ANDRICOPULO, p. 753, 2015). A técnica SBDD refere-se ao uso sistemático da estrutura do alvo macromolecular que pode ser obtido experimentalmente ou através de modelagem computacional por homologia. O método SBDD permite o planejamento de ligantes por meio da simulação intermolecular entre os ligantes e o receptor. Essa informação é aplicada aos diferentes métodos tais como a docagem molecular, que prediz a conformação do ligante com o sitio de ligação da molécula alvo. Além disso, os métodos de docagem molecular possibilitam uma predição quantitativa da energia de ligação baseada na afinidade do complexo ligante-receptor (FERREIRA et al, p. 13384, 2015). Diante do exposto, os derivados tetrahidro-1-benzazepinas figuram como uma importante classe de compostos que possuem grande diversidade biológica demonstrando atividade contra infecções parasitárias que causam a leishmaniose e a doença de Chagas, especialmente como inibidores dos alvos quinase dependente de ciclina CRK3 – dependente quinase de L. mexicana e dihidrofolato redutase de T. cruzi. Neste estudo foram utilizados 21 derivados tetrahidro-1-benzazepinas com atividades biológicas determinadas experimentalmente (PALMA et al, p. 2360, 2009) na proposição de um modelo QSAR obtido com o método dos mínimos quadrados parciais. Foi utilizada também a docagem molecular com o objetivo de avaliar as interações entre os derivados tetrahidro-1-benzazepinas e a enzima dihidrofolato redutase de T. cruzi (TcDHFR), que é essencial para o parasito e representa um alvo promissor no planejamento de novos compostos com atividade anti-tripanosoma.
Material e métodos
SELEÇÃO DE COMPOSTOS E MODELO QSAR: Para o desenvolvimento do modelo QSAR foi utilizada uma série de 21 compostos tetrahidro-1-benzazepinas com atividade biológica (IC50) contra T. cruzi reportadas no estudo de Palma et al. (2009). O conjunto consiste em dois grupos, sendo um deles o epóxi-2,3,4,5-tetrahidronafto[1,2-b]azepinas (compostos 1 a 11) e hidroxi-2,3,4,5-tetrahidronafto[1,2-b]azepinas (compostos 12 a 21). Os métodos químicos quânticos AM1, PM3 e RM1, Hartree-Fock e Teoria do Funcional Densidade (funcional B3LYP) foram utilizados para avaliar qual é mais apropriado para descrever a geometria do composto cristalográfico, 2-exo-fenil-1,4-epóxi-2,3,4,5-tetrahidronafto[1,2-b]azepina – CCDC 828009 (GONZALES et al, p. 356, 2012) e os cálculos foram realizados usando o software Gaussian 03. A avaliação do melhor método teórico para descrever a geometria deste composto foi feita por meio da análise RMS (Root Mean Square) usando o software Hyperchem 6.0, comparando-se a geometria do ligante cristalográfico àquela obtida com os métodos químico quânticos. O método mais adequado para descrever a geometria do ligante cristalográfico foi aplicado ao conjunto de 21 compostos para a otimização da geometria molecular e obtenção dos descritores moleculares do tipo eletrônicos, estruturais, espacial, topológicos, de solubilidade e geométricos obtidos a partir do software Gaussian 03, Edragon 1.0 e ALOGPS 2.1. Foi construída a matriz de dados contendo os descritores moleculares e a atividade biológica expressa como pIC50. A partir dessa matriz foi desenvolvido o modelo QSAR com o método dos mínimos quadrados parciais (PLS) usando o algoritmo de seleção de preditores ordenados – OPS (TEÓFILO, MARTINS e FERREIRA, p. 32, 2009) e o software QSAR modeling (MARTINS e FERREIRA, p. 554, 2013). DOCAGEM MOLECULAR:A docagem foi realizada predizer a conformação e as interações dos derivados tetrahidro-1-benzazepinas no sítio ativo da enzima alvo. A enzima selecionada foi a dihidrofolato redutase de T. cruzi (TcDHFR) por ser um potencial alvo no desenvolvimento de fármacos para tratar a doença de Chagas. Para essa etapa foi utilizado o software AutoDock Vina 1.1.0. A enzima foi recuperada do banco de dados PDB com o código 3KJS. A estrutura molecular TcDHFR foi tratada no programa Chimera 1.11 e consistiu na retirada do ligante cristalográfico DQ1 e das moléculas de água cristalográficas, em seguida foram adicionados os átomos de hidrogênio polares e calculadas as cargas parciais de Kollman utilizando o programa AutoDock Tools 1.5.4. Os resíduos Val26, Asp48, Ile154 e Tyr160 da enzima TcDHFR foram selecionados como flexíveis, pois são responsáveis pela ligação do ligante cristalográfico no sítio ativo da enzima (SCHORMANN et al, p. 4056, 2010). A análise das interações entre os derivados tetrahidro-1-benzazepinas e os aminoácidos da enzima TcDHFR oriundas da docagem molecular foram visualizadas no servidor PoseView.
Resultado e discussão
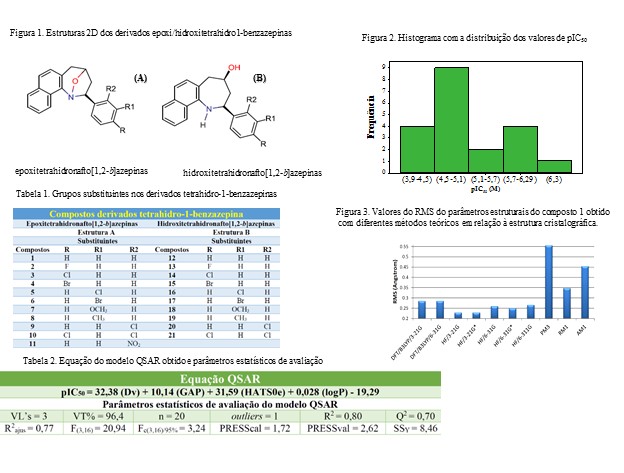
As estruturas bidimensionais dos derivados tetrahidro-1-benzazepinas são apresentadas na Figura 1 e os respectivos grupos substituintes são mostrados na Tabela 1, destaca-se que os compostos 1-11 são derivados epoxitetrahidronafto[1,2-b]azepinas, estrutura A, já os compostos 12-21 são da classe hidroxitetrahidronafto[1,2-b]azepinas, estrutura B. Os valores experimentais de IC50 expressos em µM foram convertidos para unidades de concentração molar (M) e posteriormente em pIC50 (-logIC50), pois isso garante que todas as atividades biológicas tenham valores positivos. De acordo com o histograma apresentado na Figura 2, nota-se que as atividades biológicas dos 21 compostos estão bem distribuídas considerando o intervalo de pIC50 de 3,9 a 6,3 M.
A análise do método teórico que é mais apropriado para a otimização da geometria do composto 2-exo-fenil-l-1,4-epoxi-2,3,4,5-tetrahidronafto[1,2-b]azepina (composto 1) foi feita comparando-se os desvios médios (Root Mean Square-RMS) entre a estrutura cristalográfica do composto 1 e a geometria teórica obtida com os diferentes métodos. De acordo com os valores de RMS apresentados na Figura 3, o método Hartree-Fock utilizando o conjunto de base 3-21G (HF/3-21G) foi o mais adequado na descrição da geometria do composto 1, pois foi o que apresentou o menor valor de RMS na comparação. Então, as estruturas dos demais compostos (2-21) foram construídas a partir da estrutura do composto 1 otimizada com o método HF/3-21G. Com a obtenção das geometrias otimizadas, foram determinados os descritores moleculares abrangendo as características eletrônicas, estruturais, topológicas, geométricas e de solubilidade, levando a um total de 730 descritores moleculares. O desenvolvimento do modelo QSAR foi feito em três etapas e com o uso do software QSAR modeling. Primeiro, os 730 descritores originais foram reduzidos a 265, descartando aqueles cujos valores absolutos do coeficiente de correlação de Pearson (| r |) com pIC50 foram menores que 0,35. Na segunda etapa, foi utilizado o algoritmo de Seleção de Preditores Ordenados (OPS) para a seleção de variáveis, visto que esse algoritmo consegue construir modelos por mínimos quadrados parciais (PLS) utilizando os descritores autoescalados (pré-processamento utilizado neste trabalho) e assim cada descritor tem sua importância dada com base e um vetor informativo. Como resultado do emprego do algoritmo OPS, a matriz de dados foi reduzida de 265 descritores para 40. A última etapa consistiu no refinamento da matriz de dados, realizada no software QSAR modeling e com o método OPS-PLS, pois torna-se inviável a obtenção de um modelo quantitativo com tantos descritores. Como resultado, foi obtido um modelo QSAR contendo 4 descritores como variáveis independentes e pIC50 como variável dependente, sendo eles Dv, GAP, HATS0e e logP, que representam índice de acessibilidade total, diferença entre as energias dos orbitais HOMO e LUMO, informação geométrica e topológica dos compostos, solubilidade, respectivamente. O gráfico leverage contra os resíduos de Student possibilitou identificar o composto 11 como outlier, e este foi removido do conjunto de dados. Então esse conjunto foi dividido em dois grupos, conjunto de treinamento (1, 3, 4, 6-8, 10, 12-15, 17, 18 e 20) e conjunto teste (2, 5, 9, 16, 19 e 21).
O modelo quantitativo foi obtido com o método dos mínimos quadrados parciais (PLS) e três variáveis latentes (3VL’s) e a equação matemática e os resultados dos parâmetros estatísticos de avaliação são mostrados na Tabela 2, nota-se que os quatros descritores selecionados contribuem positivamente para o valor de pIC50, sendo logP o de menor proporção. Observa-se que 3VL’s descrevem um total de 96,4% da informação original e que os descritores selecionados explicam 80,0% e predizem 70,0% da variância, representados pelos valores de R2 e Q2, respectivamente. Destaca-se que esses valores são superiores aos recomendados na literatura e que a diferença entre eles não excede o intervalo 0,2-0,3, indicando que o modelo QSAR não apresenta sobreajuste, overfitting (KIRALJ; FERREIRA, p. 770, 2009). O valor de F calculado, obtido através do teste F, é muito maior que o correspondente F tabelado com intervalo de confiança de 95%, já a soma dos quadrados dos erros de predição da validação e da calibração (PRESSval e PRESScal) são menores que soma dos quadrados dos valores de resposta (SSy), demostrando que o modelo QSAR proposto possui significância estatística (MELO; FERREIRA, p. 3577, 2009).
A robustez do modelo matemático foi avaliada pelo procedimento de validação cruzada retirando-N-compostos, utilizando-se o software QSAR modeling. Os resultados são apresentados na Figura 4, e cada barra representa a média de três repetições sendo o número máximo de compostos retirados na análise igual a 5 (N = 5). Nota-se que os valores médios de Q2 são superiores a 0,6 e com pequenas flutuações com até cinco compostos removidos, sendo os valores dos desvios-padrão menor que 0,06 e dessa forma o modelo QSAR pode ser considerado robusto (MELO; FERREIRA, p. 3577, 2009). O teste de aleatorização de y foi usado para detectar e quantificar correlações ao acaso entre a variável dependente (pIC50) e os descritores selecionados. Os resultados obtidos para todos os modelos aleatorizados são de péssima qualidade e com valores de R2 e Q2 inferiores aos do modelo real, esses resultados indicam que o modelo QSAR proposto não foi obtido ao acaso (CAMPOS; MELO, p. 19, 2014. O modelo matemático foi utilizado para predizer os valores de pIC50 dos 21 compostos estudados, foi observado que a diferença entre os valores experimentais e preditos de pIC50 em geral é pequena e menor que -0,88, exceto o para o composto 11, pois o resíduo apresentou valor igual a 4,44 confirmando a hipótese que esse composto é um outlier, porque é o único que apresenta o grupo NO2 na sua estrutura.
Os resultados da docagem molecular são apresentados na Figura 5, nota-se que os 21 compostos ficaram bem posicionados no sítio ativo da enzima dihidrofolato redutase de T. cruzi (TcDHFR), sendo que a principal interação responsável pela estabilização dos ligantes no sítio ativo da enzima é do tipo π – π (pi-pi) entre os anéis benzênicos dos compostos e o resíduo aromático Phe52, conforme mostrado na Tabela 3. Para os compostos 19 e 20, Figura 6, ocorre também a interação de hidrogênio com Ser83, sendo que essas duas interações já foram reportadas no estudo in silico de Mendoza-Martínez e colaboradores (2015), destacando a importância dessas interações. Já o composto 21 além da interação do tipo π – π, há também a interação de hidrogênio com o resíduo de aminoácido Ile154 que já foi reportado no estudo in silico realizado por Schormann e colaboradores (SCHORMANN et al, p. 4056, 2010), isso demonstra a importância dessa interação na estabilização do ligante 21. Destacam-se também na Figura 6 as interações hidrofóbicas, pois são responsáveis pela estabilização destes compostos no sítio ativo da enzima TcDHFR.

Conclusões
O conjunto de 21 compostos derivados tetrahidro-1-benzazepinas possibilitaram a proposição de um modelo matemático entre a relação quantitativa estrutura-atividade (QSAR) com capacidade explicativa e preditiva aceitáveis, robusto e que não sofre de sobre ajuste (overfitting). A técnica de docagem molecular possibilitou identificar que os compostos interagiram com resíduos de aminoácidos do sítio ativo da enzima dihidrofolato redutase de T. cruzi e que a interação do tipo π – π entre o anel benzílico dos ligantes e o resíduo aromático Phe52 da enzima TcDHFR é uma das responsáveis pela estabilização dos compostos no sítio ativo da enzima. Os compostos 19, 20 e 21 merecem destaque, pois os resultados da docagem molecular demostraram que além da interação com Phe52, eles realizam interação de hidrogênio com os outros resíduos de aminoácidos, sendo Ser83 para os compostos 19 e 20, e Ile154 para o composto 21. Portanto, os resultados obtidos neste estudo podem auxiliar na proposição e predição da atividade biológica de novos compostos tetrahidro-1-benzazepina com afinidades pela enzima dihidrofolato redutase de T. cruzi.
Agradecimentos
Os autores agradecem à Universidade Federal do Pará - UFPA
Referências
CAMPOS, J. C.; MELO, E. B. Modeling structure–activity relationships of prodiginines with antimalarial activity using GA/MLR and OPS/PLS. Journal of molecular graphics & modelling, nº 2, 19-31, 2014.
CORTES-SERRA N.; PINAZO M-J.; DE LA TORRE, L.; GALIZZI, M.; GASCON, J.; BUSTAMANTE, J. M. Diagnosis of Trypanosoma cruzi Infection Status using Saliva of Infected Subjects. The American Journal of Tropical Medicine and Hygiene, nº 98, 464-467, 2018.
DINIZ, L. F.; MAZZETI, A. L.; CALDAS, I. S.; RIBEIRO, I.; BAHIA, M. T. Outcome of E1224 – Benznidazole Combination Treatment for Infection with a Multidrug-Resistant Trypanosoma cruzi Strain in Mice. Antimicrobial agents and chemotherapy, nº 62, e00401, 2018.
FERREIRA, L. G.; OLIVA, G.; ANDRICOPULO, A. D. Target-based molecular modeling strategies for schistosomiasis drug Discovery. Future Medicinal Chemistry, nº 6, 753-764, 2015.
FERREIRA, L. G.; SANTOS, R. N.; OLIVA, G.; ANDRICOPULO, A. D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules, nº 7, 13384-13421, 2015.
GONZALES, V. E.; VIZCAYA, L. A.; MORA, A. J.; DELGADO, G. E.; BAHSAS, A.; YÉPES, A. F.; PALMA, A. Crystal Structure of 2-Exo-Phenyl-2,3,4,5-Tetrahydro-1,4-Epoxinafto[1,2]azepine. Journal of Chemical Crystallography, nº 4, 356-359, 2012.
KIRALJ, R.; FEREIRA, M. C. Basic Validation Procedures for Regression Models in QSAR and QSPR Studies: Theory and Application. Journal of the Brazilian Chemical Society, nº 4, 770-787, 2009.
MARTINS, J. P.; FERREIRA, M. C. Qsar modeling: um novo pacote computacional open source para gerar e validar modelos QSAR. Química Nova, nº 4, 554-560, 2013.
MELO, E. B.; FERREIRA, M. M. C. Multivariate QSAR study of 4,5-dihydroxypyrimidine carboxamides as HIV-1 integrase inhibitors. European Journal of Medicinal Chemistry, nº 44, 3577-3583, 2009.
MENDOZA-MARTÍNEZ, C. et al. Design, synthesis and biological evaluation of quinazoline derivatives as anti-trypanosomatids and anti-plasmodial agents. European Journal of Medicinal Chemistry, nº 3, 296-307 2015.
PALMA, A.; YÉPES, A. F.; LEAL, S. M.; CORONADO, C. A.; ESCOBAR, P. Synthesis and in vitro activity of new tetrahydronaphtho[1,2-b]azepine derivatives against Trypanosoma cruzi and Leishmania chagasi parasites. Bioorganic & Medicinal Chemistry Letters, nº 8, 2360-2363, 2009.
SACCONNAY, L.; ANGLEVIEL, M.; RANDAZZO, G. M.; QUEIROZ, M. M. F.; QUEIROZ, E. F.; WOLFENDER, J-L.; CARRUPT, P-A.; NURISSO, A. Computational Studies on Sirtuins from Trypanosoma cruzi: Structures, Conformations and Interactions with Phytochemicals. Plos One – Neglected Tropical Disease, nº 8, e2689, 2014.
SCHORMANN, N.; VELU, S. E.; MURUGESAN, S.; SENKOVICH, O.;WALKER, K.; CHENNA, B. C.; SHINKRE, B.; DESAI,A.; CHATTOPADHYAY, D. Synthesis and characterization of potent inhibitors of Trypanosoma cruzi dihydrofolate reductase. Bioorganic & Medicinal Chemistry Letters, nº 18, 4056-4066, 2010.
TEÓFILO, R. F.; MARTINS, J. P. A.; FERREIRA, M. M. C. Sorting variables by using informative vectors as a strategy for feature selection in multivariate regression. Journal of Chemometrics, nº 1, 32-48, 2009.
WIGGERS, H. J.; ROCHA, J. R.; FERNANDES, W. B.; SESTI-COSTA, R.; CARNEIRO, Z. A.; CHELESKI, J.; SILVA, A. B. F.; JULIANO, L.; CEZARI, M. H. S.; SILVA, J. S.; MCKERROW, J. H.; MONTANARI, C. A. Non-peptidic Cruzain Inhibitors with Trypanocidal Activity Discovered by Virtual Screening and In Vitro Assay. Plos One – Neglected Tropical Disease, nº 7, e2370, 2013.








