Modelagem por homologia das enzimas Histidina Amônio Liase e Urocanato Hidratase de Homo sapiens
ISBN 978-85-85905-21-7
Área
Físico-Química
Autores
Boreiko Sánchez, S. (UEPG) ; Miranda, R.R. (UEPG) ; Silva, M. (UTFPR) ; Iulek, J. (UEPG)
Resumo
Neste trabalho estão apresentadas as estruturas tridimensionais das enzimas Histidina Amônio Liase de Homo sapiens (HsHAL) e Urocanato Hidratase de Homo sapiens (HsUH) obtidas por modelagem por homologia, bem como a comparação com as proteínas homólogas de Pseudomonas putida. O diagrama de topologia mostrou a presença 58,7% hélices-α e 3,9% fitas-β para a HsHAL e 41,1% hélices-α e 12,7% fitas-β para HsUH. A sobreposição indicou que não houve grande quantidade de elementos estruturais desorganizados.
Palavras chaves
modelagem por homologia; histidina amônio liase; urocanato hidratase
Introdução
Proteínas podem ter sua estrutura tridimensional elucidada por meio das técnicas de difração de raios X, ressonância magnética nuclear (RUPP, p. 141 e 185, 2010), microscopia eletrônica (BARTESAGHI et al, p. 1, 2015), modelagem por homologia e ab initio (SANTOS FILHO; ALENCASTRO, p. 1, 2003). Estudos da enzima HsUH envolvendo casos da sua deficiência foram relatados pela primeira vez em 1971 (YOSHIDA et al, p. 305-312, 1971), seguidos por estudo posterior de dois casos descritos por Kalafatic et al, p. 1013-1019, (1980). Relatos confirmam como principais manifestações clínicas o retardo mental e distúrbios de linguagem (YOSHIDA et al, p. 305, 1971). A enzima HsHAL também está associada ao retardo mental e distúrbios de linguagem (SCRIVER; LEVY, p. 51, 1983). No Protein Data Bank (PDB) estão depositadas as estruturas da Urocanato Hidratase (1UWK) e a da Histidina Amônio Liase (1GKM) de Pseudomonas putida, organismo patógeno oportunista para os seres humanos. Assim, o conhecimento da estrutura tridimensional destas enzimas de Homo sapiens poderá contribuir a estudos de possíveis inibidores e busca por novos fármacos. Neste trabalho estão apresentados as estruturas 3D das enzimas HsUH e HsHAL obtidas por modelagem por homologia e comparação com as proteínas homólogas de Pseudomonas putida.
Material e métodos
Busca por homólogas A busca por sequências similares à da enzima alvo foi realizada com a ferramenta Basic Local Alignment Search Tool (BLAST) (PARK et al, p. 1-6, 2012). A fim de priorizar as estruturas para serem usadas como molde foi verificado o que o programa Workflow for Structural Genomic Projects (MHOLline) sugeria com o índice Blast Automatic Targeting for Structures (BATS). Ainda, os seguintes critérios foram utilizados para selecionar estruturas: porcentagem de identidade < 25%, de melhor resolução e com presença de ligantes. Sobreposição das cadeias e alinhamento das sequências de aminoácidos As estruturas tridimensionais selecionadas do PDB foram sobrepostas com a utilização do programa MultiProt (SHATSKY; NUSSINOV; WOLFSON, p. 235-250, 2002). O alinhamento entre as sequências foi obtido utilizando-se o programa ClustalW (HIGGINS; SHARP, p. 237-244, 1988) e T-coffee (NOTREDAME; HIGGINS; HERINGA, p. 205-217, 2000), a partir das estruturas sobrepostas. Modelagem por homologia Os modelos tridimensionais foram produzidos pelo programa Modeller (ESWAR et al, p. 2.9.1-2.9.31, 2006), tendo sido gerados 150 modelos. As estruturas selecionadas foram visualizadas pelo programa PyMOL (DELANO, p. 1, 2002). Validação Os modelos foram avaliados em relação à pontuação Discrete Optimized Protein Energy (DOPE) (SHEN; SALI, p. 2507-2524, 2006), GA341 (JOHN; SALI, p. 3982-3992, 2003) e pela análise do gráfico de Ramachandran com uso do programa Procheck (LASKOWSKI et al, p. 283-291, 1993). O modelo com menor valor do DOPE normalizado foi escolhido desde que estivesse de acordo com os outros critérios de avaliação estabelecidos pelo gráfico de Ramachandran e escore GA341. Sobreposição dos modelos por homologia O modelo tridimensional da enzima HsHAL obtida por modelagem por homologia foi sobreposto com a HAL de Pseudomonas putida (PDB: 1GKM) e a sobreposição da enzima HsUH foi realizada utilizando-se a estrutura homóloga (PDB: 1UWK), também de Pseudomonas putida.
Resultado e discussão
Enzima HsHAL
A ferramenta BLAST identificou 27 proteínas homólogas. Com a ferramenta MHOLline obteve-se a pontuação BATS 2 para 3 organismos diferentes, assim foram escolhidas as seguintes estruturas moldes: 1GKM, 2QVE e 3UNV com 46, 40 e 35%, de identidade sequencial.
O modelo escolhido apresentou DOPE normalizado igual a -0,880 e o gráfico de Ramachandran indicou que 91% dos aminoácidos nas regiões mais favoráveis, 7,4% em regiões adicionalmente permitidas, 0,3% em regiões generosamente permitidas e 1,4 % em regiões desfavoráveis. O modelo final está representado na figura 1-A.
Com a base de dados SCOP (CONTE et al, p. 264-267, 2002) foram explicitados os elementos das estruturas secundárias: 58,7% de hélices-α (vermelho) e 3,9% fitas-β (rosa) (Figura 1-B).
Enzima HsUH
Por meio da ferramenta BLAST, foram identificadas 6 proteínas homólogas. Com a ferramenta MHOLline de acordo com a pontuação do BATS 2, os 3 diferentes organismos foram indicados para construção do modelo. Códigos PDB: 1UWK, 2FKN e 1X87 com 33, 33 e 36% de identidade sequencial.
A estrutura escolhida apresentou DOPE normalizado igual a -0,837. O gráfico de Ramachandran indicou que 89,5% dos aminoácidos nas regiões mais favoráveis, 8,3% em regiões adicionalmente permitidas, 1,4% em regiões generosamente permitidas e 0,8 % em regiões desfavoráveis. O modelo final da estrutura da HsUH está apresentado na figura 1-D.
Com a base de dados SCOP foi possível verificar que o modelo apresentou 41,1% de hélices-α (vermelho) e 12,7% fitas-β (rosa). (Figura 1-E).
Sobreposição das homólogas com os modelos obtidos
A sobreposição mostrou que as estruturas são bastante similares em ambos os casos e, quando comparadas as estruturas de ambas enzimas homólogas foi possível constatar um deslocamento de 2 e 1 região em hélices-α (Figura 1-C e 1-F).
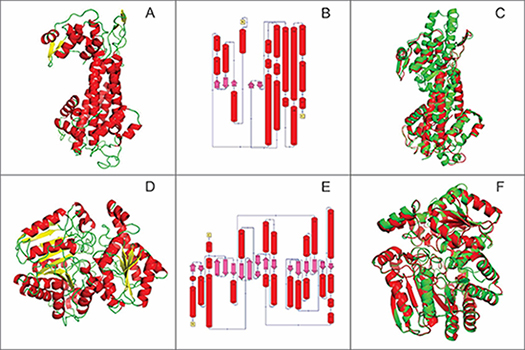
A. Estrutura da enzima HsHAL. B. Diagrama de topologia para o modelo da HsHAL. C. Sobreposição do modelo por homologia da HsHAL (verde) com a estrutura homóloga (vermelho). D. Estrutura da enzima da HsUH. E. Diagrama de topologia para a estrutura HsUH. F.
Conclusões
Pode-se concluir que os modelos gerados apresentam boa qualidade conforme os parâmetros de controle utilizados e que, quando comparados visualmente com os modelos tridimensionais das estruturas homólogas, apresentam boa sobreposição, não indicando grande quantidade de elementos estruturais desorganizados.
Agradecimentos
UEPG
Referências
BARTESAGHI, A.; MERK, A.; BANERJEE, S.; MATTHIES, D.; WU, X.; MILNE, J. L. S.; SUBRAMANIAM. 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science Express, 1-8, 2015.
CONTE L. L; Brenner S. E; HUBBARD, T. J. P.; CHOTHIA C.; MURZIN, A. G. SCOP database in 2002: refinements accommodate structural genomics. Nucleic Acids Research, 264-267, 2002.
DELANO, W. L. The PyMOL Molecular Graphics System. DeLano Scientific, 2002.
ESWAR, N.; WEBB, B.; MARTI-RENOM, M. A.; MADHUSUDHAN, M. S.; ERAMIAN, D.; SHEN, M. Y.; PIEPER, U.; SALI, A. Comparative protein structure modeling using Modeller. Current Protocols Bioinformatics, 2.9.1-2.9.31, 2006.
HIGGINS, D. G.; SHARP, P. M. Clustal: a package for performing multiple sequence alignmenton a microcomputer. Gene, no 1, 237-244, 1988.
JOHN, B.; SALI, A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Research, no 14, 3982-3992, 2003.
LASKOWSKI, R. A.; MACARTHUR, M. W.; MOSS, D. S.; THORNTON, J. M. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, no 2, 283-291, 1993.
KALAFATIC, Z.; LIPOVAC, K.; JEZERINAC, Z.; JURETIC, D.; DUMIC, M.; ZURGA, B.; RES, L. A liver urocanase deficiency. Metabolism, no 11, p. 1013-1019, 1980.
NOTREDAME, C.; HIGGINS, D. G.; HERINGA, J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology, no 1, 205-217, 2000.
PARK, Y.; SHEETLIN, S.; MA, N.; MADDEN, T.; SPOUGE, J. New finite-size correction for local alignment score distributions. BMC Research Notes, no 1, 1-6, 2012.
SANTOS FILHO, O. A.; ALENCASTRO, R. B. Modelagem de proteínas por homologia. Química Nova, no 2, 2003.
SCRIVER, C. R.; LEVY, H. L. Histidinaemia. Part I: Reconciling retrospective and prospective findings. Journal of Inherited Metabolic Disease, 51-53, 1983.
RUPP, B. Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology. 1. ed. New York: Garland Science, p. 141-733, 2010.
SHATSKY, M.; NUSSINOV, R.; WOLFSON, H. MultiProt - A Multiple Protein Structural Alignment Algorithm. In: GUIGÓ, R. e GUSFIELD, D. (Ed.). Algorithms in Bioinformatics, 235-250, 2002.
SHEN, M.; SALI, A. Statistical potential for assessment and prediction of protein structures. Protein science: a publication of the protein society, no 11, 2507-2524, 2006.
YOSHIDA, T.; TADA, K.; HONDA, Y.; ARAKAWA, T. Urocanic aciduria: a defect in the urocanase activity in the liver of a mentally retarded. The Journal of Experimental Medicine, no 4, p. 305-312, 1971.































