ISBN 978-85-85905-15-6
Área
Iniciação Científica
Autores
Gusmão, A.S. (IFBA) ; Fontineles, T.A.C. (IFBA)
Resumo
A criptolepina apresenta uma potente atividade in vitro contra o parasita da malária, além de ter sido o primeiro de uma classe de agentes intercalantes a ter uma preferência de intercalação por sítios CG do DNA por um modo de intercalação paralela e não alternada. Neste trabalho, foram usados derivados diretos da criptolepina com comprovada atividade antimalárica e obtidos por meio de estudos experimentais. Foram realizados cálculos de energia de interação empregando a metodologia DFT/DCACP, e também o funcional M062X. Os resultados mostraram que a interação entre a criptolepina e seus derivados com o DNA é bastante favorável energeticamente. Além disso, os resultados mostraram que há uma maior interação entre os pares de bases CG do DNA e os derivados contendo grupos halogenados.
Palavras chaves
criptolepina; intercalação; DFT
Introdução
O estudo teórico da interação entre biomoléculas que interagem com o DNA vêm crescendo muito nos últimos anos. Entender o mecanismo de sua atuação no processo de intercalação com o DNA é de fundamental importância no desenvolvimento de novos fármacos para o tratamento de doenças de origem genética e parasitária (MARTINEZ; CHACON-GARCIA, p.127, 2005). Recentemente, estudos experimentais descobriram uma importante classe de compostos intercalantes ao DNA devirados da criptolepina, um agente antimalárico pertencente ao grupo das indoquinolinas (LISGARTEN et. al, p.57, 2002). A criptolepina apresenta uma potente atividade in vitro contra o parasita da malária, além de ter sido o primeiro de uma classe de agentes intercalantes a ter uma preferência de intercalação por sítios CG do DNA por um modo de intercalação paralela e não alternada. Isso sugere que esse novo processo de intercalação deve levar ao desenvolvimento de novos fármacos contra a malária e o estudo teórico através de simulações computacionais proposto neste trabalho, pode auxiliar no delineamento de trabalhos experimentais. Propomos investigar as interações relevantes para descrição do processo de intercalação da criptoleptina e seus derivados potencialmente ativos contra malária, tais como 2,7-dibromocriptolepina, 7-bromo-2-clorocriptolepina, 2- bromo-7-nitrocriptolepina, dentre outros (ONYEIBOR et.al, p.2701, 2005). As simulações foram realizados por meio de cálculos baseados na Teoria do Funcional de Densidade (DFT) juntamente com a correção DCACP (LIN et. al, p.205131, 2007), do inglês Dispersion Corrected-Atom Centered Potentials e o funcional M062X para comparação. O método DCACP tem como objetivo a inclusão dos efeitos de dispersão na DFT convencional.
Material e métodos
O trabalho foi iniciado partindo-se de modelos empilhados com configurações alternadas para a interação fármaco-DNA, onde as geometrias foram obtidas da estrutura de um cristal retirado do Protein Data Bank (PDB:1K9G). Neste modelo, a criptolepina está intercalada a um dodecâmero de DNA com sítios CG assimétricos. A partir desse modelo, utilizou-se o programa Avogadro para gerar as estruturas dos derivados da criptolepina (Tabela 1) com comprovada atividade antimalárica, selecionados por meio de resultados experimentais. Usou-se o modelo empilhado para calcular as energias de interação entre os derivados interagindo com um par de base CG. Apesar de simples, o modelo envolvendo apenas um par de base e o fármaco é comumente usado na literatura especializada para estimar a energia de interação do DNA com o intercalante. As geometrias dos modelos empilhados dos derivados foram otimizadas usando o método DFT/DCACP implementado no programa CPMD e o método M062X implementado no programa Gaussian 09. Em seguida, realizou-se cálculos de energia de interação empregando a metodologia DCACP e também o funcional M062X. O cálculo das energias de interação entre a criptolepina e seus derivados e o par de base foi feito usando a equação abaixo: ∆Ecomplex=E (emp)- (E(fármaco) + E(par CG)) O funcional M062X foi utilizado devido ao seu bom desempenho em descrever energias de interação de dispersão, cuja contribuição é majoritária na interação entre moléculas intercalantes ao DNA5. Os resultados foram visualizados e analisados pelo programa de visualização GaussView.
Resultado e discussão
A Tabela 1 mostra as energias de interação calculadas para os derivados da criptolepina contendo grupos nitro, metil e halogênios na sua estrutura. Os resultados mostram que todos os derivados otimizados tanto com o método DCACP
como pelo método M062X mostraram uma energia de interação mais atrativa do que o
modelo CG-Criptolepina. Isso é condizente com os resultados experimentais obtidos
por Onyeibor et.al.
Todos os outros derivados também apresentaram energias de interação atrativas e
todas as energias mostraram-se mais estáveis frente ao modelo na configuração do
cristal (CG-Cript). Isso mostra que a maioria dos derivados da criptolepina
contendo grupos nitro ou átomos de halogênio na sua estrutura aumenta a interação
com o DNA. Os derivados contendo átomos substituintes de cloro e bromo foram mais
estáveis. O 2-bromo-7-nitrocriptolepina, o mais estável, foi cerca de 15 kcal/mol
mais estável do que o modelo CG-Criptolepina. Esses resultados indicam que deve
haver uma forte interação de halogênio significativa entre os átomos halogenados substituintes na estrutura da criptolepina com os anéis aromáticos presentes na
base CG, contribuindo para a estabilidade do complexo.
Os resultados obtidos pelo método DCACP mostraram que os derivados mais ativos
são respectivamente, o 2-bromo-7-nitro, 2,7-dibromo e o 7-bromo-2-clorocriptolepina, conforme relatado nos resultados experimentais. Entretanto,
comparando-se as energias dos dois métodos considerados, pode se observar uma
grande diferença entre as energias de alguns derivados. Isso pode ser explicado
pelo fato de o método M062X não conseguir reproduzir tão bem interações de
dispersão para moléculas contendo átomos halogenados na sua estrutura.
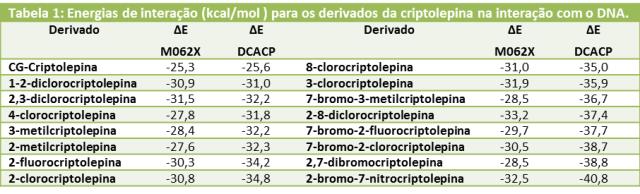
Energias de interação (kcal/mol) para os derivados da criptolepina na interação com o DNA

Geometria dos dímeros em configurações alternativas da criptolepina interagindo com um par de base CG
Conclusões
Os resultados de energias mostraram que a interação entre a criptolepina e seus derivados é bastante favorável energeticamente. Entretanto, o método DCACP foi mais condizente com resultados experimentais. Derivados que apresentam átomos substituintes de cloro e bromo na sua estrutura apresentaram energias mais estáveis quando comparado com o modelo na geometria do cristal. Isso mostra que a atividade citotóxica do fármaco em questão pode ser melhorada com a inclusão destes substituintes, tornando o fármaco mais efetivo em sua atividade antiplasmódica, conforme comprovado experimentalmente.
Agradecimentos
Os autores agradecem ao CNPq pela bolsa de pesquisa concedida, ao IFBA e ao Cenapad pelo suporte técnico para a realização das simulações.
Referências
LIN, I. C.; COUTINHO-NETO, M. D.; FELSENHEIMER, C.; VON LILIENFIELD, O. A.; TAVERNELLI, I.; ROTHLISBERGER, U. Physical Review B, nº 75, p.205131, 2007.
LISGARTEN, J. N.; COLL, M.; PORTUGAL, J.; WRIGHT, C. W.; AYMAMI, J. Nature Structural Biology ,nº 9, p.57, 2002
MARTINEZ, R.; CHACON-GARCIA, L. Current Medicinal Chemistry, nº 12, p.127, 2005.
ONYEIBOR, O.; CROFT, S. L.; DODSON, H. I.; FEIZ-HADDAD, M.; KENDRICK, H.; MILLINGTON, N. J.; PARAPINI, S.; PHILLIPS, R. M.; SEVILLE, S.; SHNYDER, S. D.; others J. Med. Chem, nº 48, p.2701, 2005
Patrocinadores








Apoio









Realização

