
Realizado no Rio de Janeiro/RJ, de 14 a 18 de Outubro de 2013.
ISBN: 978-85-85905-06-4
ÁREA: Físico-Química
TÍTULO: Estudo de modelagem molecular para compostos inibidores da acetilcolinesterase
AUTORES: Silva, A.E.S. (UNIFAP) ; Santos, C.B.R. (UNIFAP) ; Hage-melim, L.I.S. (UNIFAP)
RESUMO: A Doença de Alzheimer (DA) é a causa mais comum de demência em idosos. A progressão dos sintomas da doença se deve a modificações estruturais nas sinapses colinérgicas em determinadas regiões cerebrais e, consequentemente, à diminuição do potencial de neurotransmissão colinérgica. Para esse estudo, foram utilizadas diferentes técnicas de modelagem molecular como estratégia de planejamento de fármacos, tendo como base os inibidores de AChE descritos na literatura. O objetivo foi planejar novos potenciais inibidores desse alvo terapêutico, na tentativa de otimizar novos protótipos como futuros candidatos a fármacos para DA.
PALAVRAS CHAVES: Acetilcolinesterase; Doença de Alzheimer; Farmacóforo
INTRODUÇÃO: A Doença de Alzheimer é uma desordem neurológica devastadora e fatal, sendo que a explicação exata da causa e uma provável cura ainda são desconhecidas. O MA afeta principalmente idosos, sendo conhecido como uma doença senil e o aumento da expectativa de vida tem elevado gradualmente o número de pessoas que estão desenvolvendo a doença (RAUK, 2009).
Uma característica distintiva das anormalidades é caracterizada, microscopicamente, pela presença de placas amilóides (depósito de fragmentos de proteína beta-amilóide) e emaranhados neurofibrilares, associados à perda gradual da capacidade cognitiva e deterioração progressiva da memória e do aprendizado, resultantes de um déficit colinérgico (SHINTANI; UCHIDA,1997).
As pesquisas sobre a enzima acetilcolinesterase têm aumentado devido às novas descobertas apoiando o envolvimento da enzima na formação do peptídeo ß-amilóide durante a patogênese do Mal de Alzheimer (GUPTA; GOPI MOHAN, 2010). O objetivo deste estudo é ampliar as pesquisas na busca de novos fármacos para o tratamento de Alzheimer.
MATERIAL E MÉTODOS: A identificação da localização de um sítio de ligação numa proteína é de fundamental importância durante o planejamento de novos fármacos e, em particular, para a realização de “docking” molecular. Com esta finalidade, foi utilizado o web-servidor, Q-SiteFinder (LAURIE, 2005). O Q-SiteFinder é um método que prediz sítios de ligação favoráveis utilizando um critério puramente de energia. Esse método calcula as interações de energia de van de Waals de um grupo de prova metila com a proteína. As provas com energias de interação favoráveis são retidas e os mapas destas provas são classificadas de acordo com suas energias totais de interação. O web-servidor encontra-se disponível em http://www.modelling.leeds.ac.uk/qsitefinder/.
Em seguida, uma padrão farmacofórico comum aos inibidores selecionados foi derivado utilizando o servidor PharmaGist (SCHNEIDMAN-DUHOVNY et al., 2008). O grupamento farmacofórico candidato foi detectado por alinhamento múltiplo dos ligantes utilizados.
RESULTADOS E DISCUSSÃO: Predizer interações moleculares é o objetivo primordial da estratégia “análogo ativo” em planejamento racional de fármacos. Um modelo importante para alcançar este objetivo é o farmacóforo (DROR et al., 2004). O padrão farmacofórico derivado aos inibidores selecionados apresentou um grupo aceptor de ligação de hidrogênio e dois grupamentos aromáticos (Figura 1)
Após predizer o grupamento farmacofórico, foi identificada a localização dos sítios de ligação da enzima acetilcolnesterase através do web-servidor Q-SiteFinder, o que é importante para o planejamento de novos candidatos a fármacos. A identificação da localização de um sítio de ligação numa proteína é de fundamental importância durante o planejamento de novos fármacos e, em particular, para a realização de “docking” molecular, sendo a próxima etapa da pesquisa (Figura 2).
Figura 1
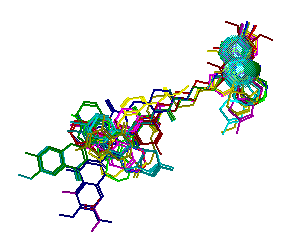
Hipótese de farmacóforo identificado pelo web servidor PharmaGist para os inibidores de acetilcolinesterase
Figura 2

Sítios de ligação preditos pelo Q-SiteFinder para a enzima Acetilcolinesterase
CONCLUSÕES: Para o tratamento da doença só são disponíveis dois grupos de drogas, o que assim como restringe bastante a evolução na busca da correção cognitiva, incentiva ainda mais pesquisadores a testarem mais drogas com o objetivo de retardar a evolução das perdas intelectuais. Com os resultados obtidos neste estudo foi possível identificar a localização dos sítios de ligação da enzima acetilcolnesterase, fato de relevância fundamental para o planejamento de candidatos a novos fármacos, além dos grupos funcionais essenciais para a interação com a enzima.
AGRADECIMENTOS: À Universidade Federal do Amapá (UNIFAP)
Ao Laboratório de Modelagem e Química Computacional (LMQC)
REFERÊNCIAS BIBLIOGRÁFICA: DROR, O.; SHULMAN-PELEG, A.; NUSSINOV, R.; WOLFSON, H. J.; Predicting molecular interactions in silico: I. A guide to pharmacophore identification and its applications to drug desing. Current Medicinal Chemistry, V. 11, nº1, p. 71-90, 2004.
GUPTA, S.; Gopi Mohan, C. 3D - pharmacophore model based virtual screening to identify dual - binding site and selective acetylcholinesterase inhibitors. Medicinal Chemistry Research (2010).
LAURIE, A. T.; JACKSON, R.M. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics, V. 21, nº 9, p. 1908-1916, 2005.
RAUK, A. The chemistry of Alzheimer”s disease. Chemical Society Reviews (2009), 38(9), 2698-2715.
SCHNEIDMAN-DUHOVNY, D.; DROR, O.; INBAR, Y.; NUSSINOV, R.; WOLFSON, H. J. PharmaGist: a webserver for ligand-based pharmacophore detection. Nucleic Acids Research, V. 36, p. W223-W228, 2008.
SHINTANI, E. Y.; UCHIDA, K. M. Donepezil: an anticholinesterase inhibitor for Alzheimer’s disease. American Journal of Health-System Pharmacy (1997), 54(24), 2805- 2810.