
Realizado no Rio de Janeiro/RJ, de 14 a 18 de Outubro de 2013.
ISBN: 978-85-85905-06-4
ÁREA: Química Orgânica
TÍTULO: ESPECTROSCOPIA DE RMN DE 1H E CÁLCULOS AB INITIO APLICADOS NA CARACTERIZAÇÃO DE COMPLEXOS ENTRE LIGANTES ORGÂNICOS E ÁCIDOS CARBOXÍLICOS
AUTORES: Bezerra de Lima, N. (UFPE) ; Ferreira da Cruz Santos, V. (UFPE) ; Karine da Luz Belarmino, M. (UFPE)
RESUMO: Neste trabalho, cálculos ab initio foram empregados para estudar as propriedades
estruturais, energéticas e espectroscópicas de complexos entre ligantes orgânicos
e ácidos carboxílicos. Nossos resultados computacionais mostraram que complexos
estáveis podem ser formados entre ligantes orgânicos (compostos nitrogenados e
compostos com grupamento sulfóxido) e ácidos carboxílicos (ácido acético e ácido
benzoico). Os cálculos GIAO revelaram que em todos os casos, a formação da ligação
de hidrogênio provocaria uma blindagem eletrônica nos núcleos de hidrogênio ácido,
este fato foi confirmado por espectroscopia de RMN de 1H para todos os casos,
exceto para o complexo entre 2,2’ bipiridina e ácido benzoico, o qual observamos
uma desblindagem eletrônica.
PALAVRAS CHAVES: RMN de 1H; cálculos ab initio; ligações de hidrogênio
INTRODUÇÃO: A identificação de sinais associados à presença de ligações de hidrogênio em
complexos entre moléculas aceptoras de próton e moléculas doadoras de próton não
é fácil através de métodos analíticos, como por exemplo, a espectroscopia de
infravermelho. Porém, a espectroscopia de RMN de 1H vem se mostrado uma
excelente técnica para comprovar a existência da ligação de hidrogênio em
complexos envolvendo ligantes orgânicos e ácidos carboxílicos[1].
Por outro lado, cálculos ab initio de orbitais moleculares podem ser utilizados
para simular computacionalmente as propriedades moleculares de interesse de
sistemas químicos, como por exemplo, as geometrias mais estáveis, estabilidades
energéticas, modos vibracionais e deslocamentos químicos dos núcleos atômicos[2-
4]. Por conta disto, esses cálculos podem ser utilizados no estudo das
propriedades de complexos envolvendo ligantes orgânicos e ácidos carboxílicos
via ligação de hidrogênio.
Neste trabalho, cálculos ab initio de orbitais moleculares foram empregados para
estudar as propriedades estruturais, energéticas e espectroscópicas de complexos
entre ligantes orgânicos (compostos nitrogenados: 2,2’ bipiridina (BIPI) e 1,10
fenantrolina (FEN) e compostos sulfóxidos: fenilmetilsulfóxido (PMSO),
dibenzilsulfóxido (DBSO) e p-tolilsulfóxido (PTSO))e diferentes ácidos
carboxílicos (ácido acético e ácido benzoico). Posteriormente, buscamos
comprovar a existência das ligações de hidrogênio nesses complexos através da
espectroscopia de RMN de 1H.
MATERIAL E MÉTODOS: Foram empregados cálculos ab initio de orbitais moleculares usando a teoria do
funcional de densidade (DFT)[5] com o funcional B3LYP[6] com o conjunto de
funções base 6-31++G(d,p) para otimização completa de geometria. Após a
otimização completa de geometria, cálculos GIAO foram realizados para simular os
deslocamentos químicos dos núcleos de hidrogênio teóricos. Todos os cálculos
computacionais foram realizados utilizando o programa de química quântica
computacional GAUSSIAN 2003.
Para a realização dos experimentos de RMN 1H, foram preparadas soluções contendo
0.2 mmol ligante orgânico e ácido carboxílico em 0,5mL de CDCl3 na proporção
1:1. Também realizamos análises das moléculas livres de complexação em solução
de CDCl3. Os experimentos de RMN de 1H foram realizados no equipamento VARIAN
UNMRS 400 MHz. Nestes experimentos utilizamos um padrão de TMS com deslocamento
químico em 0 ppm. O campo magnético (B0) utilizado foi 7 T, também foi utilizado
um pulso de 45º e Lb=5Hz.
RESULTADOS E DISCUSSÃO: A primeira etapa do trabalho consistiu na otimização completa das geometrias dos
complexos, ou seja, a simulação da geometria mais estável energeticamente, a
qual será a mais provável de existir. Em todos os casos observamos que nenhuma
frequência imaginária foi encontrada, ou seja, todos os complexos correspondem a
pontos de mínimo na curva de potencial de energia. Posteriormente foi verificado
que em todos os casos, o comprimento da ligação O-H dos ácidos carboxílicos
aumentou. Os complexos entre os ligantes orgânicos estudados neste trabalho são
estáveis energeticamente, na ordem de -42 kJ/mol para os complexos envolvendo os
compostos nitrogenados e -38 kJ/mol para os complexos envolvendo os compostos
com grupamento sulfóxido. Posteriormente, verificamos que os valores da
frequência do modo vibracional do estiramento O-H foram deslocados para menores
valores e suas correspondentes intensidades aumentadas após a complexação. Os
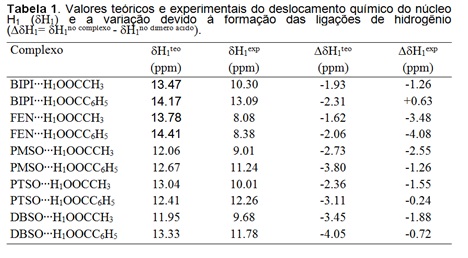
cálculos GIAO obtidos para os valores dos deslocamentos químicos dos núcleos
hidrogênios ácidos revelaram que a complexação resulta na blindagem eletrônica
desses núcleos. Estes resultados podem ser vistos na tabela 1. Através desta
tabela também podemos observar os valores experimentais obtidos via
espectroscopia de RMN de 1H. Analisando os resultados, observamos que os
efeitos da complexação no deslocamento químico dos núcleos de hidrogênio são
bastante fortes para os complexos entre compostos nitrogenados e RCOOH (R=-CH3
e –C6H5). Os resultados experimentais mostram que em todos os casos houve uma
blindagem eletrônica dos núcleos de hidrogênio ácido exceto para o complexo
entre 2,2‘ bipiridina e ácido benzoico, o qual ouve uma desblindagem, por causa
do efeito anisotrópico dos anéis aromáticos.
CONCLUSÕES: Nossos cálculos ab initio de orbitais moleculares mostraram que complexos estáveis
podem ser formados entre ligantes orgânicos (compostos nitrogenados e compostos
com grupamento sulfóxido) e ácidos carboxílicos (ácido acético e ácido benzoico).
Nenhuma frequência imaginária foi encontrada. Os cálculos GIAO revelaram que em
todos os casos, a formação da ligação de hidrogênio provocaria uma blindagem
eletrônica nos núcleos de hidrogênio ácido, este fato foi confirmado por
espectroscopia de RMN de 1H para todos os casos, exceto para o complexo entre 2,2’
bipiridina e ácido benzoico.
AGRADECIMENTOS: Os autores agradecem a PROACAD, PROPESQ, UFPE, CNPQ e FACEPE pelo suporte
financeiro e ao LQTC.
REFERÊNCIAS BIBLIOGRÁFICA: [1] Reyman D, Oliva DC, Hallwas F, Barros SMG, The Royal Society of Chemistry 1:857, 2011.
[2] Belarmino, M. K. D. L.; Lima, N. B.D., Ramos, M. N., Int. J. of Qua. Chem. 112 (2012) 3246–3251.
[3] Nascimento, R. X. D.; Belarmino, M. K. D. L.; Lima, N. B.D., Int. J. of Qua. Chem. 112, (2012) 3147–3151
[4] Lima, N. B.D.; Ramos, M. N., J. of Mol. Struct. 1008 (2012) 29–34.
[5] A.D. Becke, J. Chem. Phys. 1993, 98, 5648.
[6] P. Geerlings, F. De Proft, W. Langenacker, Chem. Rev. 2003, 103,1793.