ÁREA: Iniciação Científica
TÍTULO: CÁLCULOS TEÓRICOS DE PROPRIEDADES MOLECULARES DE COMPLEXOS DE HIDROGÊNIO FRACAMENTE LIGADOS ENVOLVENDO FTALIMIDAS E HF
AUTORES: Belarmino, M.K.L. (UFPE) ; Santos, V.F.C. (UFPE) ; Oliveira, M.S.S. (UFPE) ; Lima, N.B. (UFPE)
RESUMO: No nosso trabalho realizamos cálculos B3LYP/6-31++G(d,p) que claramente mostram que a ftalimida pode sofrer mono e dupla complexação via ligação-H. Cinco diferentes complexos foram calculados e não apresentaram nenhuma frequência negativa, o que correspondem a pontos de mínimo na curva de energia de potencial. A complexação produz mudanças significativas nas propriedades eletrônicas e vibracionais da ftalimida e do ácido fluorídrico. Em particular, a formação da ligação-H produz abaixamento de suas freqüências de estiramento para valores menores. A intensidade de estiramento HF é muito afetada pela complexação, como esperado. Diante deste trabalho, as perspectivas são realizar estudos de complexos formados entre ftalimidas e outros ácidos monopróticos lineares H-X (X= Cl, CN, Br e CCH).
PALAVRAS CHAVES: Ligação de hidrogênio; ftalimida; frequência vibracional
INTRODUÇÃO: É bem conhecido que a ftalimida (Figura 1) e seus derivados são potenciais agentes hipolipidêmicos[1] quando administrados em doses relativamente baixas de 20 mg/Kg/dia em camundongos. Uma análise dos dados experimentais indica que essa ação hipolipidêmica pode estar associada à planaridade do anel de cinco membros dessas imidas. Por exemplo, substituindo uma das carbonilas da ftalimida por um grupo –CH2 transformando-a assim em ftalimidina, produz uma redução substancial na atividade hipolidêmica. Estudos de orbitais moleculares AM1, em conjunto com técnicas estatísticas de análise multivariada, realizados por Ramos e Neto[2], permitiram melhor compreender como a mudança na estrutura molecular afeta a ação biológica dessas ftalimidas. Esses resultados revelaram que a mudança na atividade hipolidêmica pode ser explicada em termos da energia do orbital molecular mais baixo desocupado (LUMO) e pela polaridade da carbonila. Esses dados também revelaram que a forma do orbital LUMO é do tipo pi* e que quanto mais negativa for a sua energia, maior a capacidade do derivado da ftalimida em reduzir os níveis de colesterol e de triglicerídeos no sangue. O fato do LUMO ser de simetria pi* requer uma estrutura planar para uma melhor interação com o receptor, assim a quebra da planaridade torna menos negativo o LUMO e reduz assim a sua capacidade de receber carga do receptor. Isto leva a crer que esses derivados possivelmente atuam através de um processo de transferência de carga.
nosso trabalho teve como objetivo principal analisar dois aspectos:(1) o efeito da complexação na energia do LUMO e na polaridade da carbonila da ftalimida, e(2) as mudanças nas propriedades moleculares tanto da ftalimida como da espécie doadora de próton. Aqui usamos o ácido fluorídrico(HF).
MATERIAL E MÉTODOS: Para realizar esse estudo, nós empregamos cálculos de orbitais moleculares usando a teoria do funcional de densidade (DFT)[4] com o funcional B3LYP[5] com o conjunto de funções base 6-31++G(d,p). O Programa computacional usado foi o GAUSSIAN 03.
RESULTADOS E DISCUSSÃO: Nossos resultados mostram que as energias são dadas por ligação de hidrogênio (variação de energia); assim, o complexo I é o que apresenta a maior energia de estabilização por ligação-H, enquanto o complexo III é o de menor. A energia por ligação-H cai após a 2ª complexação, uma vez que a transferência de carga intermolecular diminui. Os valores de variação de energia(ZPE),BSSE mostram assim que o complexo I é mais estável que o complexo II, e na 2ª complexação, ela preferencialmente estabiliza a formação do complexo V. Os momentos de dipolo são bem distintos entre os complexos, sendo o maior deles no complexo III, 9.06 D, pela estrutura ser simétrica e pela orientação dos vetores de dipolo das duas moléculas HF, no mesmo sentido do vetor dipolo da ftalimida. O aumento de polaridade na sua formação é também significativo de 1.91 D. É interessante observar que o efeito no complexo V é exatamente o oposto do complexo, em termos da orientação dos vetores dipolos das duas moléculas de HF com relação àquele da ftalimida; seu momento de dipolo é apenas 1.10 D, e uma perda de polaridade na formação do complexo em relação á soma dos dipolos das moléculas isoladas, ou seja, sua variação dipolar é negativo (-6.05D). Como mostra a Tabela1.
A Tabela 2 apresenta dois fatores interessantes, o primeiro é que a energia do LUMO mais negativa após a formação dos complexos indica que a complexação deve contribuir para aumentar a atividade hipolipidêmica, mas o segundo fator, a redução da polaridade das carbonilas, deve diminuir o efeito na redução dos níveis de triglicerídeos, já que ele não influencia os níveis de colesterol, este apenas depende da energia do LUMO.
Os complexos
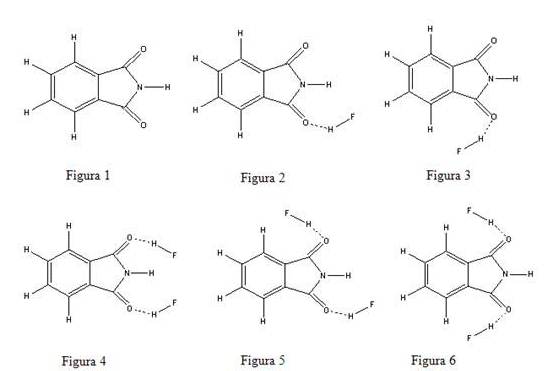
Complexos de hdrogênio obtidos envolvendo ftalimidas e HF
Tabelas

Tabelas 1 e 2
CONCLUSÕES: Nossos cálculos B3LYP/6-31++G(d,p) claramente mostram que a ftalimida pode sofrer mono e dupla complexação via ligação-H. Cinco diferentes complexos foram calculados e não apresentaram nenhuma frequência negativa, o que correspondem a pontos de mínimo na curva de energia de potencial. A complexação produz mudanças significativas nas propriedades eletrônicas e vibracionais da ftalimida e do ácido fluorídrico. Em particular, a formação da ligação-H produz abaixamento de suas freqüências de estiramento para valores menores.
AGRADECIMENTOS: Os autores agradecem a PROPESQ/UFPE, CNPQ, FACEPE e CAPES pelo suporte financeiro.
REFERÊNCIAS BIBLIOGRÁFICA: [1] (a) J.M. Chapman Jr., S.D. Wyrick, P.J. Voorstad, J.H. Maguire, G. H. Cocolas e I.H. Hall, J. Pharm. Sci., 73, (1984), 1482; (b) J.M. Chapman Jr., S.D. Wyrick, P.J. Voorstad e I.H. Hall, J., J. Med. Chem. 26 (1983) 237; (c) A.R. Murthy, S.D. Wyrick e I.H. Hall, J. Med. Chem. 28, (1985), 1591; (d) I.H. Hall, J.M. Chapman Jr. e G. H. Cocolas, J. Pharm. Sci., 70, (1981), 326.
[2] M.N. Ramos e B.B. Neto, J. Comp. Chem. 11, (1990), 559.
[3] Veja, por exemplo, (a) J.B.P. da Silva, M.R.Silva Júnior, M.N. Ramos e S.E. Galembeck, J. Mol. Struct. 744-747, (2005), 217; (b) V.H. Rusu, M.N. Ramos e J.B. P. da Silva, Int. J. Quantum Chem. 106, (2006), 2811.
[4] A.D. Becke, J. Chem. Phy. 98, (1993) 5648.
[5] P. Geerlings, F. De Proft, W. Langenacker, Chem. Rev., 103, (2003), 1793.
