ÁREA: Físico-Química
TÍTULO: ESTUDO TEÓRICO DA INTERAÇÃO ENTRE OXIGÊNIO MOLECULAR E NANOCLUSTERS Pt-Cr.
AUTORES: VARELA JÚNIOR, J. J. G. (IQSC/USP) ; BRITO, J. G. L. (UFMA) ; SILVA, A. L. P. (UFPB) ; COSTA, H. R. (UFMA) ; TANAKA, A. A. (UFMA) ; SILVA, A. B. F. (IQSC/USP)
RESUMO: Este trabalho apresenta um estudo aplicando o funcional B3LYP, para estudar a estrutura
eletrônica de nanoclusters de platina dopados com Cr e suas interações com oxigênio
molecular. As análises das populações de Mulliken e de NBO para a interação entre
oxigênio molecular e os nanoclusters Pt2 e Pt-Cr revelaram que ocorre transferência de
carga dos orbitais s e d dos metais para os orbitais p do oxigênio, resultando no
preenchimento dos orbitais antiligantes da molécula de oxigênio, provocando a quebra da
ligação OO e formação de ligações hibridizadas MetalO. As curvas de superfície de
energia potencial revelaram um valor de energia de dissociação da ligação OO em 1,0 eV
sobre o cluster Pt2 e esse valor decresce para 0,56 eV para o cluster Pt-Cr.
PALAVRAS CHAVES: nanoclusters de platina, teoria do funcional da densidade, adsorção de oxigênio molecular.
INTRODUÇÃO: As reações eletroquímicas envolvendo o oxigênio, em particular a reação de redução de
oxigênio, continuam a despertar o interesse dos eletroquímicos por envolverem
complexidades cinéticas, pela necessidade de melhores eletrocatalisadores e pela
importância destas reações em sistemas de conversão de energia eletroquímica, além de
sínteses químicas, processos biológicos e de corrosão. Dessa forma, Balbuena et al.
[1] realizaram estudos da redução de oxigênio em ligas de Co, Ni e Cr, embutidos em
matrizes de Pt, usando a metodologia B3PW91/LANL2DZ e postularam que Cr e Co podem
atuar como sítios ativos para a dissociação de O2 em vez de serem oxidados como
sítios de sacrifício e identificaram XPt e XXPt (X= Cr e Co) como sítios mais ativos
para promover a dissociação de O2. Gobal et al. [2] estudaram a adsorção de oxigênio
atômico e molecular sobre nano clusters PdxCu3-x (x=0-3) neutros e carregados
negativamente usando a metodologia B3PW91/LANL2TZ(f), onde foi observado que as
formas e as energias de adsorção dependem fortemente da carga e da composição dos
clusters. O Modo de Adsorção mais estável para oxigênio molecular sobre todos os
clusters neutros foi o modelo ponte, com oxigênio adsorvido sobre os sítios Pd-Cu,
com energia de adsorção -103,7 kJ/mol, enquanto que sobre os clusters carregados
negativamente o modelo ponte sobre os sítios Pd-Pd, com energia de adsorção -140,9
kJ/mol e sobre Cu-Cu, com energia de adsorção -172,9 kJ/mol, foram mais estáveis.
Logo, diante do exposto, o objetivo deste trabalho é realizar um estudo aplicando a
Teoria do Funcional da Densidade (DFT), utilizando o funcional B3LYP, para estudar a
estrutura eletrônica de nanoclusters de platina dopados com Cr e suas interações com
oxigênio molecular.
MATERIAL E MÉTODOS: Este estudo foi conduzido por meio de cálculos de estrutura eletrônica baseados na
teoria do funcional da densidade. A otimização das estruturas, além dos cálculos de
energia de ligação, distribuição de cargas e freqüências vibracionais dos
nanoclusters de platina dopados com alumínio e sua interação com oxigênio molecular,
foram realizadas com a utilização do programa GAUSSIAN 03, Revision B04 [3]. Os
cálculos de DFT foram realizados utilizando o funcional híbrido de três parâmetros de
Becke com as correções de gradiente fornecidas pelos funcionais de Lee, Yang e Parr
(B3LYP) [4,5]. As funções de bases LANL2DZ (Los Alamos National Laboratory 2-double-
z, usada para os átomos Pt e Cr), 6-311G** (usada para o átomo O). A presença de
polarização na camada d pode assegurar uma possível transferência de carga do
substrato para o adsorbato. Os cálculos de frequências vibracionais das estruturas
otimizadas foram usados para gerar as energias do ponto zero (ZPE), entalpias (H),
entropias (S), energias livres de Gibbs (G) e as correções do ponto zero. A avaliação
da estrutura eletrônica foi feita através do método NBO (Natural Bond Orbitals), que
localiza os orbitais canônicos e os transforma em orbitais de centro, orbitais de
ligação, orbitais de caroço e de pares isolados, de acordo com a visão de estrutura
química de Lewis.
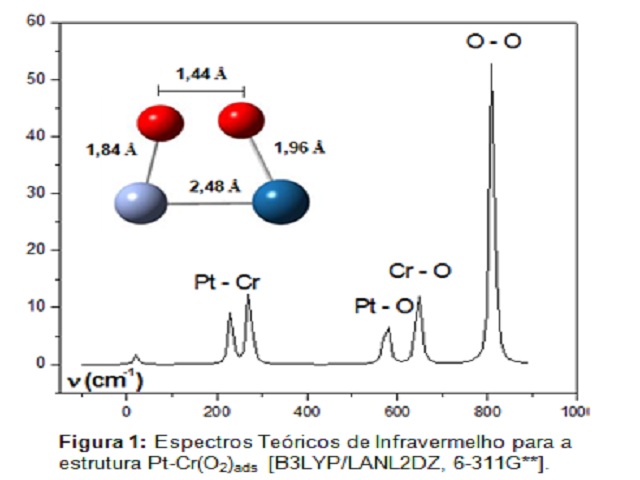
RESULTADOS E DISCUSSÃO: Os resultados obtidos mostraram que para a estrutura Pt-Cr(O2)ads o estado
fundamental é quinteto e a adsorção de oxigênio molecular ocorre seguindo o modelo
ponte, acompanhada de dissociação da molécula de oxigênio. Podemos observar na Figura
1 que os átomos de oxigênio estão ligado assimetricamente aos átomos de Pt e Cr, com
distância PtO igual a 1,96 Å e ângulo O-Pt-Cr de 69,7º enquanto que a distância CrO
encontrada foi de 1,84 Å com ângulo O-Cr-Pt igual a 78,5º. Após a adsorção, a
distância interatômica OO passa a ser de 1,44 Å, com freqüência vibracional de
811,61 cm-1 e com carga de Mulliken igual a -0,392 para o átomo de oxigênio ligado ao
Cr e -0,315 para o átomo de oxigênio ligado a Pt. A Tabela 1, apresenta as cargas
atômicas de Pt, Cr e O, e resultados das análises populacionais de Mulliken para a
estrutura Pt-Cr(O2)ads e para efeito de comparação as populações dos orbitais das
estruturas antes da interação também foram colocadas. Podemos observar um decréscimo
na população dos orbitais s e d da Pt e do Cr e consequentemente as populações dos
orbitais p dos átomos de oxigênio aumentaram, de forma assimétrica, resultando em
cargas atômicas diferentes entre os átomos de oxigênio ligados com os diferentes
metais. Sendo assim, podemos inferir que elétrons são transferidos dos orbitais s e d
da platina e do cromo para os orbitais p do oxigênio. Essa transferência resulta no
preenchimento dos orbitais antiligantes (π*) da molécula de oxigênio, causando a
quebra da ligação OO e formando as ligações hibridizadas: PtO com 32% de
contribuição de Pt(1)[6s(24%) + 5dx2-y2(75%)] e 68% de O(4)[2 (6%) + 2px(94%)]; e Cr
O com contribuições de 25% para Cr(2)[4s(20%) + 3dx2-y2(80%)] e 75% de O(3)[2s(4%) +
2px(96%)].

CONCLUSÕES: As análises das populações de Mulliken e de NBO revelaram que ocorre transferência de
carga dos orbitais s e d dos metais para os orbitais p do oxigênio, provocando a quebra
da ligação OO e formação de ligações hibridizadas Metal O. Curvas de superfície de
energia potencial revelaram um valor de aproximadamente 1,0 eV para a barreira de
dissociação da ligação O O para oxigênio molecular adsorvido em Pt2. Sobre Pt-Cr
esse valor decresce para 0,56 eV, mostrando que a dissociação de oxigênio molecular
ocorre mais facilmente sobre Pt-Cr que sobre o cluster Pt2.
AGRADECIMENTOS: Os autores agradecem à FAPEMA, ao CNPQ, ao IQSC/USP e a UFMA.
REFERÊNCIAS BIBLIOGRÁFICA: [1] BALBUENA, P. B.; ALTOMARE, D.; AGAPITO, L.; SEMINARIO, J. M., Theoretical Analysis of Oxygen Adsorption on Pt-Based Clusters Alloyed with Co, Ni, or Cr Embedded in a Pt Matrix, Journal of Physical Chemistry B, v.107, p.13671-13680, 2003.
[2] GOBAL, F.; ARAB, R.; NAHALI, M. A comparative DFT study of atomic and molecular oxygen adsorption on neutral and negatively charged PdxCu3-x(x=0-3) nano-clusters. Journal of Molecular Structure: THEOCHEM, v.959, p. 15-21, 2010.
[3] FRISCH, M. J. et al. Gaussian 03, Revision B.04; Gaussian, Inc.: Pittsburgh PA, 2003.
[4] BECKE, A. D., Density-functional Exchange-energy aproximation with correct asymptotic behavior., Physical Review A, v. 38, p. 3098-3100, 1988.
[5] LEE, C.; YANG, W.; PARR, R.G.; Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Physical Review B, v.37, p.785-789, 1988.
