ÁREA: Iniciação Científica
TÍTULO: Estrutura Eletrônica de Nitretos de Ferro Substituídos PdFe3N e Pd3FeN
AUTORES: PADILHA, G. (URI) ; ARAÚJO, M. M. (URI) ; PAVEGLIO, G. C. (URI) ; SANTOS, A. V. (URI) ; NARDES, M. (URI)
RESUMO: Deve-se entender fisicamente as propriedades do estado fundamental das ligas e, assim, calcular a estrutura eletrônica dos materiais pesquisados. Neste trabalho foram feitos cálculos de estrutura eletrônica utilizando o método FPLAPW (Aproximação Linear de Ondas Planas) comparando com o método LMTO (Método Linear de Orbitais Muffin-Tin). Assim, fez-se o estudo das ligas PdFe3N e Pd3FeN, onde encontrou-se para o PdFe3N o parâmetro de rede de 3,8755 x 10-10 m e 3,7708 x 10-10 m na fase ferromagnética e paramagnética, respectivamente. A liga Pd3FeN apresentou 4,1237 x 10-10 m e 4,0913 x 10-10 m como parâmetro de rede na fase ferromagnética e paramagnética, respectivamente. Baseado nisso, também é discutido a pressão crítica, módulo de Bulk, densidade de estados (DOS) e momento magnético.
PALAVRAS CHAVES: estrutura de bandas; fplapw.
INTRODUÇÃO: Devido à evolução tecnológica dos últimos tempos, existe uma crescente busca por novos e melhores materiais. Sendo assim, estuda-se cada vez mais ligas com diversas características. Mais especificamente a pesquisa de compostos à base de nitretos metálicos ganha força devido à sua vasta área de aplicação na indústria metal mecânica. Por exemplo, os metais de transição quando ligados ao nitrogênio têm como principal característica a capacidade de dar resistência ao desgaste e a corrosão, sendo foco de diversos artigos tanto experimentais como teóricos [1-5]. As propriedades dos nitretos de ferro tornam-se muito diferentes quando substituímos um átomo de ferro por um átomo de outro metal de transição [6], mudando as propriedades do estado fundamental dos nitretos metálicos e transformando sua estrutura de bandas e magnética, que serão os alvos principais do nosso estudo.
Faz-se uso da interpretação dos orbitais Muffin-Tin, aplicando os conceitos tanto físicos como químicos, também utiliza-se recursos computacionais. Para tanto, faz-se um modelo de célula unitária utilizando uma estrutura FCC (cúbica de face centrada).
Escolheu-se esta estrutura baseado em estudos experimentais de ligas do tipo Fe1-xPdxFe3N, onde temos a informação que o paládio aloja-se preferencialmente no corner da estrutura [6, 7]; a fim de comparar os resultados é proposta também uma estrutura onde o paládio se aloja na face.
Neste trabalho faz-se o uso de métodos de primeiros princípios a fim de entender-se o comportamento de algumas propriedades físicas do estado fundamental dos nitretos de ferro substituído PdFe3N e Pd3FeN.
MATERIAL E MÉTODOS: Utilizou-se o método All-Electron Full Potential Linear Augmented Plane Wave (FPLAPW) method [8]. Os cálculos são baseados na teoria funcional da densidade com Aproximação do Gradiente Generalizado (GGA) para o potencial da troca-correlação [9]. O raio das esferas de Wigner-Seitz foi tomado como 2,0 Å para o átomo de Pd e 1,70 Å para os demais átomos no nitreto PdFe3N e, 1,80 Å para o Pd3FeN em todos os átomos. Dentro das esferas de Wigner-Seitz, as densidades de carga e os potenciais foram tomados como as funções de onda e, são expandidos nos termos dos harmônicos esféricos, tendo o momento angular (lmax) igual a 10 para as funções de onda, e 6 para as densidades dos potenciais de carga. Para assegurar a convergência para a zona de Brillouin, devida a integração, foram utilizados 1000 k-pontos, empregando o método melhorado do tetrahedron [10]. O número de ondas planas aumentadas incluídas é de aproximadamente 80 por átomo, que corresponde a RmtKmax = 8. Para calcular a densidade de estados usou-se 1500 k-pontos na zona de Brillouin inteira. O critério de convergência é uma faixa de energia em torno de 2,18 x 10-25 J. Nesse trabalho será discutido o módulo de Bulk, a estrutura magnética do sistema, bem como as modificações causadas pela inclusão do átomo de paládio na matriz do nitreto de ferro γ-Fe4N. Os resultados obtidos serão comparados com estudos já realizados para esta estrutura, onde foi utilizado o método LMTO (Linear Muffin-Tin Orbital) de Andersen, com Aproximação de Esferas Atômicas (ASA), nas próximas seções.
RESULTADOS E DISCUSSÃO: Ao analisar os dados obtidos na tabela 1, observa-se que a liga Pd3FeN tem um parâmetro de rede maior que a liga PdFe3N, devido o tamanho do átomo de Pd ser maior que os demais, e esse nitreto possuir três átomos de Pd na célula unitária. Mas em ambos os casos o parâmetro de rede aumentou em relação ao nitreto γ-Fe4N [11], seguindo a mesma tendência de cálculos realizados pelo LMTO [1,3].
O módulo de Bulk é definido como a pressão necessária para que ocorra uma variação no volume, o PdFe3N apresenta um valor elevado em relação a outros nitretos [1], sendo, portanto, esse nitreto mais resistente quando submetido à pressão, resultado da introdução do átomo de N no centro da estrutura. Ao fazer-se a comparação com os valores encontrados para outros nitretos [7], o valor do módulo de Bulk é menor do que o valor encontrado em cálculos utilizando o método LMTO.
A pressão crítica é definida como a pressão necessária para que ocorra uma transição entre a fase ferromagnética e paramagnética. O nitreto Pd3FeN apresentou uma pressão crítica muito maior em relação ao outro nitreto estudado, mas esse valor é intermediário em relação a outros nitretos estudados utilizando o método LMTO [7].
Quanto à magnetização das ligas, observa-se que o átomo de paládio apresenta um momento magnético maior quando está situado no corner, e não na face, mostrando que a magnetização é afetada pela posição que o átomo ocupa na célula unitária, ocorrendo a formação de uma nova estrutura eletrônica, e não apenas uma média entre os momentos magnéticos. Também pode-se observar que o PdFe3N apresenta uma magnetização maior que o Pd3FeN, em parte devido haver três átomos de ferro nesse nitreto. Na figura 1 fica evidente o magnetismo dos nitretos, já que ocorre uma diferença entre os estados up e down.
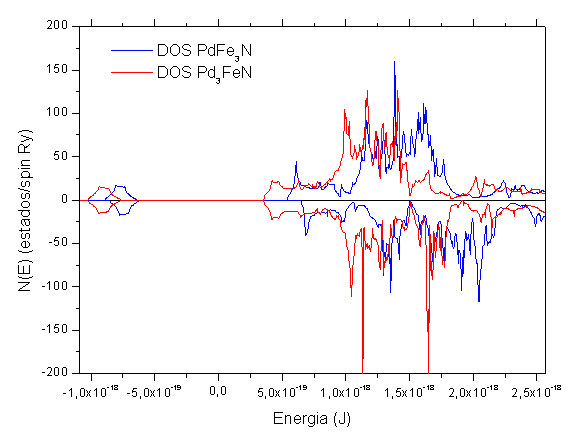

CONCLUSÕES: Primeiramente observa-se uma expansão da célula do γ-Fe4N com a introdução do átomo de paládio, o que era esperado, pois esse átomo tem um raio iônico maior que o do ferro. Observa-se também que o momento magnético no nitreto PdFe3N é maior que no nitreto Pd3FeN. Nos dois casos a magnetização no sítio do ferro aumentou em comparação com o γ-Fe4N. Em relação à densidade de estados, observa-se que no nitreto PdFe3N ela foi promovida para energias mais altas em comparação ao outro nitreto e, pode-se observar que o desemparelhamento de elétrons é menor no Pd3FeN.
AGRADECIMENTOS: Ao Núcleo de Tecnologia de Informação da URI Campus Santo Ângelo e aos demais colegas do GIESS (Grupo Interdisciplinar de Estado Sólido e Simulação).
REFERÊNCIAS BIBLIOGRÁFICA: [1] Kuhnen, C. A.; dos Santos, A. V.; J. Alloys Compd. 2004, 384, 80.
[2] Matar, S.F.; Demazeou, G; Hagenmuller, P.; Armitage, J. G. M.; Ried, P.C.; Eur. J. Solid State Inorg. Chem. 1989, 26, 517.
[3] Kuhnen, C.A.; de Figueiredo, R.S.; dos Santos, A.V.; J. Magn. Magn. Mater. 2000, 219, 58.
[4] Hashizume, S; Chino, A; Sato, K; Sakai, J.I.; Matsushima, I.; Soc for the Promotion of Science. 1996, 45, 90.
[5] dos Santos, A. V.; Kuhnen, C. A.; Krause, C .A.; Phys. B. 2006, 382, 290.
[6] Cordier-Robert, C.; Foct, J.; Eur. J. Solid State Inorg. Chem. 1992, 29, 39.
[7] Kuhnen, C. A.; dos Santos, A. V.; J. Alloys Compd. 2000, 297, 68.
[8] Blaha, P.; Schwarz, K.; Sorontins, P.; Trickey, S. B.; Comput. Phys. Commun. 1990, 59, 399.
[9]. Perdew, J.P.; Burke, S.; Ernzerhof, M.; Phys. Rev. Lett. 1996, 77, 3865.
[10] Blochl, P.E.; Jepsen, O.; Andersen, O. K.; Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 49, 16223.
[11] Cottenier, S. et al. Phys. Status Solidi B. 2009, 246, 909.
