ÁREA: Iniciação Científica
TÍTULO: Cálculos de Primeiros Princípios da Estrutura Magnética de Nitretos de Ferro Substituídos de Manganês
AUTORES: PAVEGLIO, G. C. (URI) ; PADILHA, G. (URI) ; MONÇALVES, M. A. (URI) ; NARDES, M. (URI) ; SANTOS, A. V. DOS (URI)
RESUMO: No intuito de projetarmos materiais com maior dureza, resistência a corrosão e outras propriedades utilizadas na indústria química e metal mecânica, realizamos o cálculo da estrutura de bandas do estado fundamental de nitretos de ferro substituídos. Desta forma, substituímos um átomo de ferro por um átomo de manganês, transformando-se o gama-Fe4N no MnFe3N, mas para uma comparação, também calculamos a estrutura de banda do Mn3FeN. Utilizando o método LAPW, calculamos a fase paramagnética, ferromagnética e antiferromagnética, onde determinamos volume de equilíbrio e energia de formação no equilíbrio. Observamos que há uma forte ligação entre a localização do sítio com a magnetização, sendo que o sítio do córner apresentou uma magnetização maior do que os outros sítios.
PALAVRAS CHAVES: banda, estrutura, nitreto de ferro subtituído de manganês
INTRODUÇÃO: Nos últimos anos, muito se tratou do nitreto gama-Fe4N [1,2], porém sabe-se que a introdução de um metal na matriz deste nitreto provoca várias alterações na sua estrutura eletrônica e, consequentemente, em algumas propriedades macroscópicas [3,4]. Os nitretos de ferro substituídos vêm sendo alvos de trabalhos tanto experimentais quanto teóricos [5,6]. Devido as suas propriedades, os nitretos de ferro substituídos são utilizados na indústria metal mecânica e química industrial, bem como o seu estudo teórico proporciona evolução nos métodos de cálculo de estrutura eletrônica e física e química do estado fundamental de sólidos. Neste trabalho, vamos calcular a estrutura de bandas de nitretos de ferro substituídos de manganês, obtendo as propriedades do estado fundamental.
Para tanto, utilizamos o método LAPW, utilizando Aproximação do Gradiente Generalizado (GGA), proporcionando resultados precisos dentro e fora das esferas de Wigner-Seitz [7]. Modelamos o nosso cálculo utilizando a célula unitária FCC, sendo que o nitrogênio situa-se no centro da célula, o ferro no centro das faces e o manganês no córner da célula, para a liga MnFe3N. Para comparação, calculamos a liga Mn3FeN, em que o nitrogênio encontra-se no centro da célula, o manganês no centro das faces e o ferro no córner da célula.
Após a realização dos cálculos das fases paramagnética, ferromagnética e antiferromagnética para as duas ligas, obteremos o volume de equilíbrio e seremos capazes de discutir suas propriedades básicas, tais como magnetização, densidade de estados e transferência de carga, sendo que isto será demonstrado no decorrer do trabalho.
MATERIAL E MÉTODOS: Determinar a estrutura eletrônica dos sólidos envolve considerar um número infinito de férmions interagentes. Em um cálculo real, somos levados a efetuar algumas aproximações, como por exemplo, a aproximação de um elétron. Neste caso, como primeiro passo, resolvemos a equação de Schrödinger para um elétron movendo-se em um campo médio dos outros elétrons mais o campo dos núcleos. Trabalhos mais recentes usando métodos de primeiros princípios, como os realizados por Blügel e outros, usando FLAPW (Full Linear Augmented Plane Waves) apresentam uma boa concordância com trabalhos experimentais realizados por Celinski [6] e outros. Este método tem como base a teoria do gradiente generalizado. Sendo assim dividimos o espaço colocando esferas de Wigner-Seitz em volta dos átomos onde o potencial é esférico simétrico. Utilizando um valor de 1.7 como raio das esferas do ferro, manganês e nitrogênio, realizamos o cálculo do potencial no espaço intersticial destas esferas. Deste modo, resolvemos a equação de Schrödinger para um potencial inicial, utilizando o processo de Hartree-Fock para obtermos o potencial final. Determinando este potencial calculamos a função de onda e, desta forma, podemos obter as propriedades do estado fundamental. Tal teoria traz uma importante ferramenta para os físicos e químicos, pois podemos com ela obter informações, não somente local, mas, por ser um gradiente, é possível adquirirmos informações com um alcance maior.
RESULTADOS E DISCUSSÃO: Realizamos o cálculo do nitreto MnFe3N, encontrando o valor de parâmetro de rede no equilíbrio a = 3.6731 x 10-10 m, e -2.1937 x 10-14 J como sendo a energia total de formação no equilíbrio e, B = 296.8121 x 109 Pa, com sendo o módulo de Bulk, para a fase paramagnética; a = 3.7322 x 10-10 m, e -2.1937 x 10-14 J, B = 228.3348 x 109 Pa, para a fase ferromagnética; a = 3.7177 x 10-10 m, e -2.1937 x 10-14 J, B = 227.7586 x 109 Pa, para a fase antiferromagnética. Em seguida, calculamos a liga Mn3FeN e encontramos o valor de parâmetro de rede no equilíbrio a = 3.6785 x 10-10 m, e -2.0942 10-14 J, B = 277.1132 x 109 Pa, para a fase paramagnética; a = 3.8100 x 10-10 m, e -2.0942 x 10-14 J, B = 118.7348 x 109 Pa, para a fase ferromagnética; a = 3.7322 x 10-10 m, e -2.0942 10-14 J, B = 242.1431 x 109 Pa, para a fase antiferromagnética. Conforme demonstrado na tabela abaixo, encontramos o valor de 4.82 µB e 4.7887 µB na magnetização por célula unitária, para o composto MnFe3N, na fase ferromagnética e antiferromagnética, respectivamente. Também observamos que o átomo que estava situado no sítio do córner apresentou maior magnetização que os outros sítios presentes, demonstrando assim uma forte conexão da localização do sítio com a magnetização. Este acontecimento é explicado pelo fato do átomo que está situado no sítio do córner perder mais carga do que os outros átomos, ajustando a estrutura da célula e, consequentemente, apresentando uma magnetização maior. Temos uma perda de carga para o sítio intersticial pois notamos que há a diminuição da carga na célula unitária, diminuindo assim o raio atômico dos átomos, proporcionando um maior empacotamento dos nitretos. Este fato demonstra que houve a formação de uma nova estrutura magnética no gama-Fe4N com a introdução de Mn.
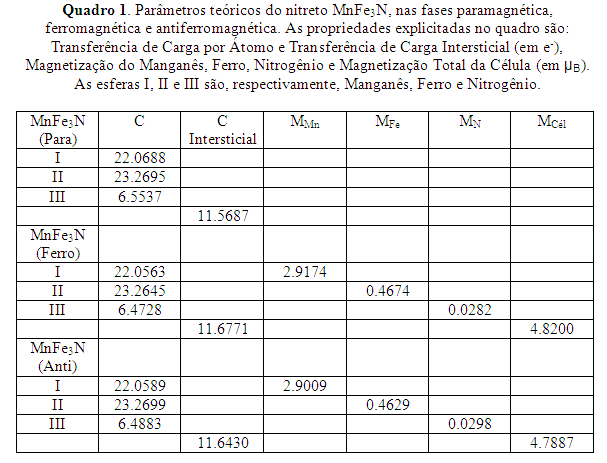

CONCLUSÕES: Observamos que o átomo que está situado no córner apresentou uma magnetização maior do que nos outros sítios. Esta magnetização maior é decorrente do ajuste de estrutura na célula unitária, necessário devido à perda de carga dos outros sítios. A magnetização dependerá da posição do sítio e também qual átomo estará nesta posição pois o trabalho apresentou uma diferença de magnetização com a utilização de diferentes átomos na mesma posição. Com relação a magnetização, todos os nitretos calculados apresentaram uma magnetização menor que a magnetização experimental encontrada para o gama-Fe4N.
AGRADECIMENTOS: Ao Núcleo de Tecnologia de Informação da URI-Santo Ângelo e aos demais colegas do GIESS (Grupo Interdisciplinar de Estado Sólido e Simulação).
REFERÊNCIAS BIBLIOGRÁFICA: 1. Kunhen, C., Figueiredo, R.S. de, Drago, V., and Da Silva, E. Z., Journal of Magnetism and Magnetic Materials 111, 95 (1992).
2. Peltzer y Blancá, E. L., Desimoni, J., Christensen, N. E., Emmerich, H., Cottenier, S. Phys. Status Solidi B 246, No. 5, 909-928 (2009)/DOI 10.1002/pssb.200844401.
3. Kunhen, C., dos Santos, A. V. Journal of Alloys and Compounds 384 (2004) 80-87.
4. Krause, J. C. Tese de Doutorado, Universidade Federal do Rio Grande do Sul, Brasil, 2000.
5. Atiq, S., Ko, H. S., Siddiqi, S. A. and Chin, S. C., Appl. Phys. Lett. 92, 222507 (2008).
6. Z. Celinski, B. Heinrich, J. F. Cochran, W. B. Muir, A. S. Arrot and J.
Kirschner, Phys. Rev. Lett., 65, 1156 (1990).
7. Perdew, J. P., Chevary, J. A., Vosko, S.H., Jackson, K. A., Pederson, M. R., Singh, D. J. and Fiolhais, C. (1992). Phys. Rev. B 46, 6671.
