ÁREA: Materiais
TÍTULO: Modificações na Estrutura de Bandas do Ni3Al Causadas Pela Introdução de Um Átomo de Carbono Intersticial
AUTORES: A. V. DOS SANTOS (URI)
RESUMO: Ao utilizarmos métodos teóricos de cálculos de primeiros princípios, podemos ter um melhor entendimento das propriedades do estado fundamental. Assim, estudamos os efeitos da inserção de um átomo de carbono na estrutura do Ni3Al, mudando a sua estrutura cristalina FCC para uma estrutura perovskite da forma Ni3AlC. Inicialmente, mostramos o parâmetro de rede no equilíbrio dos dois compostos, onde temos uma boa concordância com o valor experimental. Apresentamos também, a estrutura magnética onde encontramos uma baixa magnetização no composto Ni3AlC e uma magnetização apreciável no composto Ni3Al. Para obtenção destes resultados utilizamos o método the Linearized Augmented Plane Wave (LAPW) method com Aproximação de Gradiente Generalizado (GGA).
PALAVRAS CHAVES: nitretos, lapw, bandas
INTRODUÇÃO: A fim de obtermos novas combinações de materiais, devemos compreender do ponto de vista da teoria química e física as propriedades dos mesmos. Na tentativa de entendermos essas propriedades desses novos materiais, neste trabalho calculamos a estrutura de bandas dos compostos Ni3Al e Ni3AlC. Nossa principal motivação é saber quais as alterações provocadas pela inclusão de um átomo de carbono na matriz do composto Ni3Al, originando a estrutura perovskites Ni3AlC. Resultados experimentais atestam que esta transformação é possível e os artigos teóricos tratam principalmente de compostos na estrutura Ni3Al (WANG, 2004). Assim o objetivo desse trabalho recai em verificarmos as propriedades físicas relacionadas a estrutura eletrônica do novo composto formado, comparando com o composto anterior. Na literatura temos alguns artigos que utilizam esta metodologia de compararmos alguns resultados teóricos com experimentais, para nitretos de ferro substituídos (FIGUEIREDO et al., 2001), em relação ao parâmetro de rede, momento magnético e propriedades hiperfinas. Para realizarmos os cálculos de estrutura de bandas utilizamos o Full Potential Linear Augmented Plane Wave (FPLAPW). Como resolvemos a estrutura eletrônica, podemos estudar as propriedades químicas, utilizando as funções de ondas, obtidas através de um processo cíclico do tipo Hartree-Fock, tendo as funções de onda podemos inferir sobre as propriedades do estado fundamental, que serão demonstradas no decorrer do trabalho, assim, modelando este nitreto.
MATERIAL E MÉTODOS: A compreensão da distribuição dos elétrons nos sólidos permite o entendimento de algumas de suas propriedades físicas. Isto implica em dizer que o problema básico consiste em calcular os estados estacionários para um sistema de elétrons interagentes movendo-se em um campo eletrostático periódico, originado pelos núcleos fixos, ou seja, obter-se a estrutura eletrônica dos sólidos. Quando nos referimos a núcleos fixos estamos usando a aproximação Born-Oppenheimer, isto é, tratamos os elétrons e núcleos separadamente. Isto possibilita o cálculo da energia do estado fundamental como função das posições dos núcleos, a qual pode ser usada em um estágio posterior como energia potencial para o movimento dos núcleos.
Determinar a estrutura eletrônica dos sólidos envolve considerar um número infinito de férmions interagentes. Num cálculo real, somos levados a efetuar algumas aproximações, como por exemplo, a aproximação de um elétron. Neste caso, como primeiro passo, resolvemos a equação de Schrödinger para um elétron movendo-se em um campo médio dos outros elétrons mais o campo dos núcleos. Este campo médio é determinado pela distribuição da carga eletrônica, mais correção por efeito de correlação e troca, sendo normalmente calculado de maneira auto-consistente, usando a teoria de HartreeFock.
Os cálculos são baseados na teoria do funcional da densidade com a aproximação do gradiente generalizado (GGA) com potencial de correlação e troca (PERDEW et al., 1996). O raio das esferas de Wigner-Seitz foi tomado como 1.70 Å para todos os átomos. Dentro das esferas de Muffin-Tin, as densidades da carga, os potenciais e as funções de onda são expandidos nos termos dos harmônicos esféricos. Para assegurar a convergência para a zona de Brillouin , 1000 k-pontos foram usados.
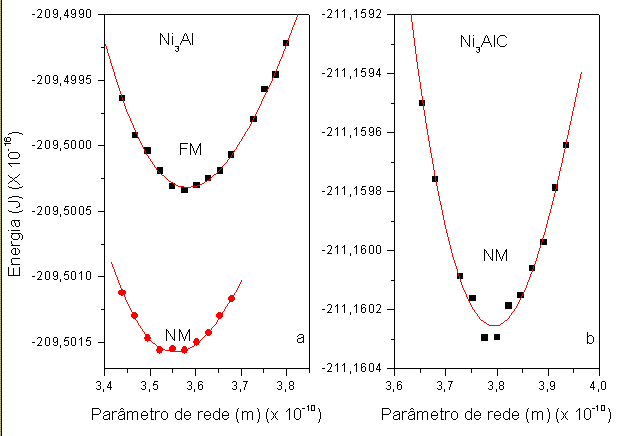
RESULTADOS E DISCUSSÃO: Inicialmente, começamos a discutir a energia de formação. Na figura 1 a e b temos a energia de formação em função do parâmetro de rede. Notamos que existe um mínimo de energia, com isto concluímos que esta fase FCC de ambos os compostos existe, como experimentalmente está mostrado nas referências (TONG et al., 2006; ZHONG et al., 2005). Temos um parâmetro de rede de 3,5828 x 10-10 m para a fase ferromagnética do Ni3Al, concordando com o parâmetro de rede experimental 3,5661 x10-10 m. Comparando com a fase ferromagnética, observamos uma boa concordância dos nossos cálculos com a experiência. Já em relação ao carbeto Ni3AlC, encontramos um parâmetro de rede de 3,7915 x 10-10 m, concordando com o estudo experimental (TONG et al., 2006); vemos que a medida que aumentamos a percentagem de carbono, existe um aumento do parâmetro de rede, mostrando assim a mesma tendência experimental. Através de cálculos da estrutura eletrônica, podemos avaliar o modulo de Bulk dos nitretos, onde encontramos para a fase ferromagnética do Ni3Al 173,98 GPa e para a fase não magnética 186,01 GPa. A introdução do átomo de carbono faz com que o modulo de Bulk aumente para 202,74 GPa, estes resultados ainda não foram verificados experimentalmente para que possamos compará-los. Notamos que existe momento magnético no sítio do Ni, confirmando os resultados experimentais (ZHONG et al., 2005), também concordando com resultados teóricos encontrado utilizando o método LMTO-ASA (WOJNECKI et al.,2004). Já no composto Ni3AlC, encontramos um momento magnético muito próximo de zero, também encontrado experimentalmente (ZHONG et al., 2005), onde a medida que aumentamos a concentração de C diminui o momento magnético no sítio do Ni.
CONCLUSÕES: Após a realização dos cálculos de estrutura de bandas dos compostos, podemos obter algumas informações preciosas sobre os compostos e testar o método LAPW, que mostrou uma razoável concordância com as propriedades experimentais. O parâmetro de rede teórico encontrado foi muito próximo do experimental nos dois casos. Quanto a magnetização, existe um momento magnético no sítio do Ni no caso do Ni3Al, e um momento magnético muito baixo no caso do Ni3AlC, também concordando com a experiência. Desta forma, temos resultados bastante importantes sobre a estrutura eletrônica destes compostos.
AGRADECIMENTOS: URI - Núcleo de Tecnologia da Informação(N.T.I.)
REFERÊNCIAS BIBLIOGRÁFICA: D. ZHONG; J.J. MOORE; E. SUTTER; B. MISHRA. Surface and Coatings Technology. 200 (2005) 1236 1241.
J.P. PERDEW; S. BURKE; M. ERNZERHOF. Physical Review Letters. 77 (1996) 3865.
P. TONG; X.B. ZHU; B.C. ZHAO; R. ANG; W.H. SONG; Y.P. SUN. Physica B. 371 (2006) 6367.
R. WOJNECKI; K. LAWNICZAK-JABLONSKA; J. KACHNIARZ; R.C.C. PERERA. Journal of Alloys and Compounds. 362 (2004) 189197.
R.S. DE FIGUEIREDO; J. FOCT; A.V. DOS SANTOS; C.A. KUHNEN. Journal of Alloys and Compounds. 315 (2001) 42.
Y. WANG; Z. K. LIU; L. Q. CHEN. Acta Materialia. 52 (2004) 26652671.
