ÁREA: Química Orgânica
TÍTULO: Determinação da barreira de rotação interna da ligação N-C(O) em N-arilamidas e N-arilcarbamatos terciários por cálculos computacionais usando teoria do funcional de densidade
AUTORES: GRASEL, F. S. (UFRGS) ; NETZ, P.A. (UFRGS) ; FONTOURA, L.A.M. (ULBRA)
RESUMO: Neste trabalho, avaliou-se, por cálculos B3LYP/6-31G(d,p), a diferença da barreira rotacional da ligação N-C(O) entre N-metilacetanilida e N-fenil-N-metilcarbamato de t-butila e alguns derivados p-substituídos (OCH3, CH3, Cl, Br, CN, NO2), comparando os resultados com dados experimentais. A análise dos resultados indicou que as acetanilidas possuem o valor de barreira rotacional superior aos N-fenilcarbamatos. Outro resultado importante é o efeito dos substituintes no anel aromático. Grupos doadores de elétrons ligados diretamente ao anel provocaram um aumento da barreira rotacional. Grupos retiradores causaram um efeito inverso. Todas as barreiras rotacionais estimadas através do cálculo B3LYP/6-31G(d,p) são bastante próximas das experimentais citadas na literatura.
PALAVRAS CHAVES: barreira rotacional, amidas, carbamatos
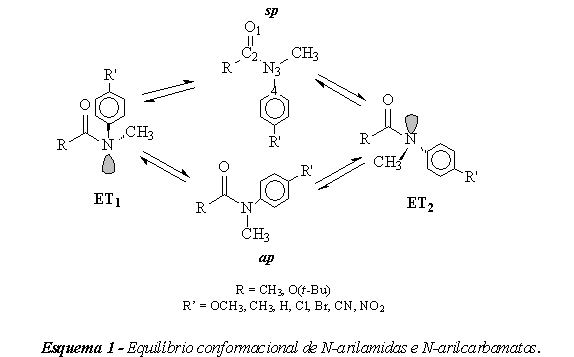
INTRODUÇÃO: Amidas são alvos de estudo particularmente importantes uma vez que suas propriedades interessam a áreas que vão da bioquímica à ciência de materiais. Basta observar que a ligação N-CO está presente em polipetídeos naturais como as proteínas, e sintéticos, como as poliamidas. Carbamatos, apesar da variada atividade biológica, têm sido relativamente menos estudados que as amidas. Em geral, estas classes de moléculas apresentam duas geometrias de equilíbrio, resultado da ressonância do par de elétrons isolado do nitrogênio com a carbonila. Ao processo de interconversão de uma geometria de equilíbrio na outra está associada uma barreira energética chamada de barreira rotacional (Esquema 1). O mecanismo da rotação desta ligação é influenciado por fatores eletrônicos e estéreos, sendo aos primeiros o principal foco deste trabalho. Neste aspecto, amidas e carbamatos terciários derivados da anilina são substratos particularmente interessantes, uma vez que efeitos eletrônicos podem ser avaliados através de substituintes apropriados no anel ou na carbonila. Uma evidência do equilíbrio conformacional é obtida por H1-RMN. Os espectros desta classe de moléculas apresentam sinais duplicados, resultado do equilíbrio entre as duas geometrias, mas que coalescem com o aumento da temperatura. Desta forma, este trabalho visa estimar, através de modelagem molecular, as barreiras rotacionais do N-metilacetanilida e N-fenil-N-metilcarbamato de t-butila e alguns derivados p-substituídos. Os valores calculados foram comparados com dados experimentais da literatura (SMITH ET AL., 2004; Yamasaki ET AL., 2003).
MATERIAL E MÉTODOS: Os compostos estudados são apresentados na tabela 1. Foram realizados cálculos semi-empíricos pelo hamiltoniano AM1 e cálculos de funcional de densidade B3LYP no conjunto de bases 6-31G(d,p) contidos nos programas Pc Spartan Pro versão 1.0.5 (Wavefunction, 2000) e Gaussian 98 (Gaussian, 1998) respectivamente. Foram realizadas as otimizações de geometria para as diversas conformações de cada composto pelo método semi-empírico AM1 para ângulos de diedro C4-N3-C2-O1 restritos de 0 (rotâmero sp) a 360º (rotâmero ap) com incrementos de 10º (Esquema 1). Além disso, as conformações de equilíbrio foram procuradas por otimização das conformações obtidas para diedros de 0 e 180º porém liberados da restrição de diedro. As geometrias obtidas na otimização por AM1 com energias mínimas foram utilizadas como input para o cálculo de Funcional de Densidade no conjunto de bases B3LYP/6-31G(d,p) sem qualquer restrição de diedro. As duas geometrias de máxima energia, tomadas como aproximações dos estados de transição (ET1 e ET2), também foram utilizadas como input respeitando-se as respectivas restrições de diedro. As barreiras de rotação interna foram estimadas como as diferenças entre as energias das geometrias de equilíbrio e dos estados de transição.
RESULTADOS E DISCUSSÃO: As estimativas das barreiras de rotação interna são apresentadas na tabela 1. Na média, as barreiras encontradas para amidas terciárias por cálculo de Funcional de Densidade são 1,2 kcal mol-1 superiores às obtidas para carbamatos terciários. Isto se deve ao fato que o grupo t-butóxi do carbamato é melhor doador de elétrons que o a metila da amida, permitindo uma menor participação do nitrogênio na ressonância com a carbonila. Para avaliar o efeito de R, é necessário discutir, antes, o efeito do grupo arila sobre o nitrogênio. O grupo arila de amidas e carbamatos compete pelo par de elétrons do nitrogênio, fazendo com que as barreiras sejam menores do que em análogos alifáticos. O efeito é mais pronunciado quando a arila apresenta um grupo retirador de elétrons, como o grupo nitro. Do outro lado, os derivados p-metoxilados apresentam a maior barreira. No caso destes dois grupos estudados, o efeito do substituinte em posição para pode afetar em até 2,7 kcal mol-1 aproximadamente no valor da barreira rotacional. Os cálculos DFT/B3LYP estimam as barreiras cerca de 5 % inferiores aos dados experimentais (SMITH ET AL., 2004; Yamasaki ET AL., 2003) para amidas e até 5% e 3 % para os carbamatos.

CONCLUSÕES: As barreiras foram estimadas entre 10,5 e 14,4 kcal mol-1, com valores menores para os carbamatos. Grupos doadores de elétrons ligados ao grupo arila provocam um aumento na barreira e o efeito contrário é obtido quando o temos um grupo retirador de elétrons. As barreiras rotacionais estimadas através do cálculo B3LYP/6-31G(d,p) são bastante próximas das experimentais citadas na literatura.
AGRADECIMENTOS:
REFERÊNCIAS BIBLIOGRÁFICA: SMITH, B. D.; GOODENOUGH-LASHUA, D. M.; DSOUZA, C. J. E.; NORTON, K. J.; SCHMIDT, L. M.; TUNG, J. C. 2004. Substituent effects on the barrier to carbamate C-N rotation. Tetrahedron Letters, 45: 2747-2749
YAMASAKI, R.; TANATANI, A.; AZUMAYA, I.; SAITO, S.; YAMAGUCHI, K.; KAGECHICA, H. 2003. Amide conformational switching induced by protonation of aromatic substituent. Organic Letters, 5: 1265-1267
